A few Books.
The war on Ivermectin. The Medicine that could have ended the pandemic, by Pierre Kory.
The Real Anthony Fauci. Bill Gates Big Pharma and the Global war on Democracy and Public Health, by Robert F Kennedy Jr.
A Plague upon our House, by Scott Atlas. Scott Atlas served as a member of the White House Coronavirus Task Force and Special Advisor to President Trump. He relates an insider's view of events at the White House.
Latest Videos.
Joe Rogan interviews Robert F. Kennedy Jr. (Complete, Unedited Interview). 238M.
Tucker Carlson; Kennedy is winning. 24M.
Robert Kennedy; Endless war abroad brings violence to the Unites States. 20M.
Tucker Carlson & Mike Pence; Ukraine persecutes Christians. 12M.
Tucker Carlson & Mike Pence; Tanks for Ukraine, but nothing for you. 6M.
Here are a few videos concerning the Ukraine war and its causes. These express views that are much closer to the truth than the fairy tales the evil media tells you.
Ray McGovern (former CIA head of Russia desk) explains the U.S. role in the 2014 Ukraine coup. 40M.
Prof. John Mearsheimer (University of Chicago) states that NATO is totally U.S. run. 2M.
Mearsheimer predicts the (remarkedly foolish) U.S. policy will lead to Ukraine being wrecked (2015). 2M.
Mearsheimer looks at the causes and consequences of the 2014 Ukraine coup (2015). 93M.
Mearsheimer looks at the causes and consequences of the 2022 Ukraine war (2022). 74M.
India Today interviews Sergei Lavrov (the Russian Foreign Minister). 72M.
Gideon Rose (Editor Foreign Affairs magazine) on the Colbert Report 02/24/2014. 23M. Funny, in a tragic way.
Putin speaking at the Valdai International Discussion Club, 27 Oct. 2022. 277M 3:38:44 Full speech.
This Tucker Carlson interview with Todd Wood is amazing. 67M. Here are a few quotes:
"The Ukraine war is not about freedom, its not about Russia, even. Its about the deep-state wanting to protect this,... whatever you want to call it,... I call it the safe space for organized crime, where they can do anything they want to do, with no transparency, with no accountability,..."
"Zelensky is run by Kolomoyskyi, who is a Jewish oligarch who also pays for all the Azov & Pravyi sektor (neo-Nazi) battalions in the east." ... "the ones with all the swastikas,..."
My question is; Why are Jews like Joseph Biden, and Ihor Kolomoyskyi, funding Nazis?
It appears that in 2014 a group of religious Jews took over Ukraine. Instead of sticking to the well tried method of infiltrating both sides of the parliament, they threatened the elected leader, Yanukovych, who fled to Russia, and they assumed power in a violent coup. This was a mistake, as Ukrainians in the east mistook this coup for a neo-Nazi takeover, and refused to acknowledge the coup government as legitimate. This led to the civil war. If they had stuck to their old play-book they would probably now control all of Ukraine, and few would have noticed, or been able to do anything about, the change in power.
It is not clear who threatened Yanukovych but it was likely Tyahnybok's crowd. Tyahnybok himself is likely a Jew as he
1) is the leader of a neo-Nazi party,
2) conspired with the Jew Victoria Nuland (U.S. Under Secretary of State for Political Affairs), and
3) has won all (nine, or ten) court cases bought against him for inciting ethnic hatred.
It is notable that the 2014 coup government found positions for the Jew Arseniy Yatsenyuk (Prime Minister of Ukraine, 27 February 2014), the Jew Petro Poroshenko (President of Ukraine, 7 June 2014) and the Jew Vitali Klitschko (Mayor of Kiev, 25 May 2014) but no position was found for the (supposed) neo-Nazi Oleh Tyahnybok (Svoboda Party leader) when he lost his position in the parliament some months later (October 2014). Subsequently, the Jew Volodymyr Groysman (14 April 2016) became Prime Minister and the Jew Volodymyr Zelensky (20 May 2019) became President. So much for the fable of neo-Nazi's taking over Ukraine in a right-wing coup. The neo-Nazis just provided a smoke screen for the Jew takeover.
The Ukraine gambit appears to be an attempt to replay the second world war.
1933: We have a Jew takeover of Germany disguised as a Nazi takeover.
The takeover occurred when the Jew Hitler assumed dictatorial powers.
Leads to war against the Soviet Union.
2014: We have a Jew takeover of Ukraine disguised as a neo-Nazi takeover.
The takeover is already complete (except in the east).
Leads to war against Russia.
Seeing that many of the politicians in "neo-Nazi" Ukraine are Jews, or of Jewish descent, I wondered if the same may have been true in Nazi Germany. So, I checked the surnames of a large number of people from the Nazi era. What I found is truly amazing. Note that all surnames designated Jewish can be found in the books on Jewish Genealogy by Alexander Beider, Lars Menk, and Emanuel Elyasaf.
Adolf Hitler [Jew surname].
Below is a list of Nazi Field Marshalls.
Looks like they were all of Jewish descent.
Fedor von Bock [Jew surname].
Werner von Blomberg [Jew surname].
Walther von Brauchitsch = Brauch-itsch [Jew surname-son of]
Ernst von Busch [Jew surname].
Hermann Göring = Gör-Ring [Jew surname-Jew surname] Luftwaffe.
Wilhelm Keitel [Jew surname].
Albert Kesselring = Kessel-Ring [Jew surname-Jew surname] Luftwaffe.
Ewald von Kleist [Jew surname].
Gunther von Kluge [Jew surname].
Georg von Küchler [Jew surname].
Wilhelm Ritter von Leeb [variant of Jew surname Lieb?]
Wilhelm List [Jew surname].
Erich von Manstein [Jew surname]. Born Lewinski [Jew surname].
Erhard Milch [Jew surname] Luftwaffe.
Walther Model [Jew surname].
Friedrich von Paulus [variant of the Jew surname Paul?]
Walther von Reichenau [Jew surname]
Erwin Rommel [variant of Jew names Frommel and Trommel?]
Gerd von Rundstedt = Rund-stedt [Jew surname-town]
Ferdinand Schörner [variant of the Jew surname Tschorne?]
Hugo Sperrle = Sperr-le = [Jew surname-common suffix] Luftwaffe.
Erwin von Witzleben [variant of Witzler-ben? Jew surname-son of]
Those of Jewish descent in Hitler's life (non-military).
Martin Bormann [Jew surname]. Hitler's private secretary. Money man.
Eva Braun [Jew surname]. Hitler's mistress and wife.
Hans Frank [Jew surname]. Hitler's lawyer.
Ulrich Graf [Jew surname]. Hitler's personal companion 1920-1923.
Gertrud Junge [Jew surname] nee Humps. Hitler's private secretary.
Theodor Morell [Jew surname]. One of Hitler's personal physicians.
Karl Brandt [Jew surname]. One of Hitler's personal physicians.
Albert Speer [Jew surname]. Hitler's personal architect and city planner.
Fritz Wiedemann [Jew surname]. Personal adjutant to Hitler.
Julius Schreck [Jew surname]. Hitler's chauffeur.
Karl von Frank [Jew surname]. Hitler's genealogist.
Hugo Blaschke [Jew surname]. Hitler's dentist.
Hugo Erlanger [Jew surname]. Hitler's landlord for ten years.
Some other Nazi leaders.
Adolf Eichmann [Jew surname]. Head of the Scientific Museum for Jewish Affairs.
Rudolf Hess [Jew surname]. Deputy to the Fuehrer.
Heinrich Himmler [Jew surname]. Leading National Socialist politician.
Alfred Rosenberg [Jew surname]. Leading proponent of National Socialist ideology.
Julius Streicher [Jew surname]. National Socialist politician.
Wilhelm Messerschmitt [Jew surname]. Aircraft designer and manufacturer.
Hjalmar Schacht [Jew surname]. Financier, president of the Reichsbank.
Joseph Goebbels [variant of the Jew surname Göbel?] National Socialist politician and propagandist.
Reinhard Heydrich = Heyd-Rich [Jew surname-Jew surname] Administrator of the concentration camps.
Here is a list of the Nazi Chiefs of General Staff.
They were probably all of Jewish descent.
Heinz Guderian [Jew surname]. Chief of General Staff Jul 1944 to Mar 1945
Adolf Heusinger [Jew surname] Chief of General Staff Jun 1944 to Jul 1944
Kurt Zeitzler [variant of the Jew name Weitzler?] Chief of General Staff Sep 1942 to Jul 1944
Franz Halder [variant of the Jew name Halde?] Chief of General Staff Sep 1938 to Sep 1942
Ludwig Beck [Jew surname] Chief of General Staff Jul 1935 to Aug 1938
Various leaders.
იოსებ ჯუღაშვილი [variant of the Jew name ჯუდაშვილი] (Joseph Stalin) General Secretary of the Soviet Union; ჯუდაშვილი translated means "son of Judah".
Mikhail Gorbachev [variant of the Jew name Gorbaczyński?] President of the Soviet Union
Boris Yeltsin [variant of the Jew name Peltson?] President of Russia
Angela Merkel [Jew surname] nee Kasner [Jew surname] Chancellor of Germany
Olaf Sholtz [variant of the Jew name Shultz?] Chancellor of Germany
Annalena Baerbock = Baer-Bock [Jew surname-Jew surname] German Foreign Minister
Adolf Hitler [Jew surname] Chancellor of Germany
Franklin Roosevelt [variant of the Jew name Rosenfelt] President USA
George Bush [variant of the Jew name Busch?] President USA
Donald Trump [variant of the Jew name Trumpf?] President USA
Lech Kaczyński [Jew surname] President of Poland
Jarosław Kaczyński [Jew surname] Prime Minister of Poland
Mateusz Morawiecki [variant of the Jew names Morawiec Morawietz etc?] Prime Minister of Poland
Stepan Bandera [variant of the Jew surname Bander?] Ukrainian Nationalist
Volodymyr Zelensky [Jew surname] President of Ukraine
Volodymyr Groysman [Jew surname] Prime Minister of Ukraine
Denys Shmigal [variant of the Jew name Schmigelski] Prime Minister of Ukraine
Vitali Klitschko [variant of the Jew name Klitsch?] Mayor of Kiev
Mark Rutte [Jew surname] Dutch Prime Minister
Klaus Johannis [Jew surname] President of Romania
Giorgia Meloni [variant of the Jew name Melon?] Italian Prime Minister
Kaja Kallas [variant of the Jew name Challasch?] Estonian Prime Minister
Petr Pavel [Jew surname] Czech President; Chief of the NATO Military Committee
Jaap Scheffer [Jew surname] NATO Secretary General 2004-2009
Jens Stoltenberg [variant of the Jew name Stoltzenberg?] NATO Secretary General 2014-
Charles Michel [Jew surname] European Council President
Ursula von der Leyen [Jew surname] nee Albrecht [Jew surname] President of the European Commission
Here is a list of Jewish surnames taken from the following books:
Handbook of Ashkenazic Given Names and Their Variants by Alexander Beider
A Dictionary of Jewish Surnames from Galicia by Alexander Beider
A Dictionary of German-Jewish Surnames by Lars Menk
Handbook of Ashkenazic Given Names and Their Variants by Alexander Beider
A Dictionary of Jewish Surnames from Italy, France and "Portuguese" Communities by Alexander Beider
A Dictionary of Jewish Surnames from Maghreb, Gibraltar, and Malta by Alexander Beider
The Jacobi Papers: Genealogical Studies of Leading Ashkenazi Families by Paul Jacobi & Emanuel Elyasaf
General Surovikin and Prigozhin; Traitors.
The Russian General Surovikin (and associates) organized the withdrawal from Kherson (Ukraine).
The main reason Surovikin ordered Russian forces to withdraw was that the Russians had just comprehensively stopped the Ukrainian Kherson offensive. All was relatively quiet. Any counter-offensive would have punched a hole right through the Ukrainian lines, their forces would have been encircled, and destroyed. It was imperative that the Russians were pulled back before the precarious state of the Ukrainian forces became known.
At this time the Ukrainians were way overextended and were not able to fight this two front war. One front had to be closed down. As the front east of the Dnieper river was by far the stronger, Surovikin had the Russians abandon the front west of the Dnieper. To make sure that the Russians could not easily reopen this front, Surovikin had the southern bridges over the Dnieper blown up. That's right, Surovikin blew up the bridges useful to the Russians. Surovikin never touched the bridges that were of vital importance to the Ukrainians. In fact, the northern bridges over the Dnieper are all still standing.
Surovikin claims that the 30,000 Russian soldiers west of Kherson could not be supplied with the necessary ammunition, etc. This of course is total garbage, considering that they had just done exactly this in stopping the Ukrainian Kherson offensive. Surovikin's claim is simply a lie. Remember, back in February the whole 30,000 man army, with all its equipment, had crossed to Kherson within a couple of days. Similarly, when they withdrew. Thus in the weeks where the civilians were pulled out of Kherson, the 30,000 man army could have been heavily reinforced, and supplied with enough equipment to smash the Ukrainian forces, but, of course, this was not wanted.
Since there were no good excuses for pulling out of Kherson, Surovikin had to grasp at straws, suggesting that artillery/missile strikes could cause the sudden breaking of the Kakhovka dam, which would wash away the bridges, and trap the 30,000 man army west of the Dnieper. Of course, even thousands of hits on the dam would not result in catastrophic failure. A gradual uncontrolled leakage of water may have occurred, resulting in a short-term flooding of some areas but nothing that would wash away bridges. (And even if the bridges were lost it would be easy enough to resupply the men by landing craft, ferries, and other boats. Resupply by aircraft would likely be too dangerous.)
Another reason that the Russians were ordered out of Kherson is that the Ukrainians had no way to stop a determined advance toward Odessa. Without Kherson as a staging area the Russians would no longer have this option. If the Russians had tried to advance on Odessa they would have met minimal resistance. The reason the Ukrainians have to continually tell you how strong their forces are, is because they are not strong.
It is clear that as long as Surovikin (and associates) are in charge, there will be no Russian attempt to cut Ukrainian supply lines as the Ukrainian army would collapse in short time. The talk of a winter offensive to cut the supply lines is just talk. It will not happen as it would quickly lead to a Ukrainian defeat.
Surovikin will continue to claim that attacking into the teeth of the Ukrainian defenses is the only way to save Russian lives. As the Ukrainians sit in their concrete bunkers and mow down any exposed Russian forces, Surovikin will tell us that things have to be done this way, to save Russian lives. Any talk of finding one of the thousands of weak spots in the Ukrainian line, breaking through the line there, and attacking the concrete bunkers from the back, will be taboo. Since frontal attacks require wasting huge quantities of ammunition such attacks will probably continue till the Russians exhaust their supplies. The best tactic for destroying fortified positions will never be allowed.
And then Putin will give Surovikin a medal.
The last section was written before the Wagner group actually sought out weak spots in the Ukrainian line, and broke through there, with the intention of encircling their enemy, and, in the absence of surrender, attacking from the back. It is now clear that the winter offensive, with all its advantages, will not occur. The Russian high command is clearly waiting for the Ukrainian side to receive thousands more anti-tank weapons before any major offensive to cut supply.
If the Russian High Command controlled the Wagner Group they would have been ordered to withdraw already. And the Ukrainians would have staged another miraculous counter offensive. Just like in Kherson and east of Kharkov. But since the Russian High Command does not control the Wagner Group they will force them to withdraw by cutting off their ammunition. Then the traitors will hand the Ukrainians yet another miraculous counter offensive.
Prigozhin has said: "As for the supply of ammunition to the Wagner PMC, Akhmat, some other units have shared what they could. Generals who are around the leadership of the Russian military grouping sign documents for which afterwards they can be put in a jail cell by the Military Prosecutor’s Office, just to provide us [PMC Wagner] with ammunition, which we get directly from the wheels. At the same time, the Chief of General Staff and the Minister of Defense are giving orders to not only stop providing PMC Wagner with ammunition, but not even help us with air transport. Now I received information that they had even cancelled distribution of sapper shovel, so the guys could entrench themselves up. There is simply a direct opposition, which is called nothing else but an attempt to eliminate the Wagner PMC. This can be considered equal to treason against the Motherland. This comes at a time when the Wagner PMC is fighting for Bakhmut, losing hundreds of its fighters every day."
Some days later, Prigozhin said in a message posted on his Telegram channel, "In order to stop me from asking for ammunition, they turned off all special [government] phone lines in all of the offices and [Wagner] units ... and blocked all [my] passes to the agencies responsible for making decisions," He subsequently called on ordinary Russians to help him put pressure on the country's regular army to share their supplies of ammunition with Wagner fighters. "Now I can only ask [for more supplies] through the media and... most likely will be doing just that," Prigozhin said.
It seems that Prigozhin frustrated the Russian High Command's effort to save Bakhmut, and they are not happy. The traitors have plans to restructure the Wagner group into something more pliable. A group that will withdraw when told to withdraw.
In Feb 2023 it was reported that the Russians were only 3.8 km from physically cutting the only road in and out of Bakhmut. But this never happened. Why? This short video describes the state of the encirclement on 19 Feb 2023. In the video it was predicted that the battle for Bakhmut would be over in a few days. However, in late February, or early March, the fighting to complete the encirclement stopped completely. At this time all roads in and out of Bakhmut came under Russian fire control.
No one seems to know why the encirclement was halted.
Have the Russian traitors stopped the fighting so that the chosen can walk out and fight another day?
The preferred option was probably for the traitors to order a withdrawal allowing the Ukrainians to stage another miraculous counter offensive, just like in Kherson and east of Kharkov.
However, the Wagner Group refused to cooperate, so they cut off it's ammunition.
But even that did not work as Prigozhin was still able to get ammunition from Akhmat, and a few other units.
Eventually Prigozhin was reduced to calling favors that enabled him to get ammunition without it being processed directly through the Russian military. So the traitors took away his phones.
Since March 1, due to lack of ammunition, the Wagner group has not tried to close the encirclement of Bakhmut.
Prigozhin's promise that the Wagner group would take Bakhmut, in spite of the Russian MOD, was premised on his being able to find ammunition elsewhere. It appears that he was not able to do so. Thus his troops are wasting their time fighting for the small village of Orikhovo-Vasylivka, far to the north-west of Bakhmut.
How little interest the Russian High Command has in winning the war can be seen in the fight for the village of Orikhovo-Vasylivka. One notes that the main road from Chasiv Yar (Bakhmut) to Slavyansk passes only 3 kms from Orikhovo-Vasylivka and that the two are separated only by open fields and an irrigation canal that parallels the road. Not only that, but this road forks to Kramatorsk only 5 kms away. Yet no member of the Russian High Command has suggested to Prigozhin that he should have his men cut the roads. Perhaps Prigozhin should have done this himself, but he seems to have a problem with ammunition.
In recent days Prigozhin seems to have come to some arrangement with the Russian High Command whereby he gets the ammunition he needs as long as he doesn't use it to encircle Bakhmut. Of course, this arrangement will lead to the unnecessary death of hundreds of Russians, but this doesn't seem to be much of a concern. The unnecessary deaths being those incurred storming Bakhmut rather than encircling it and forcing a surrender (i.e., starving them out).
And, of course, many more Russian lives were lost when Prigozhin announced Wagner would take no prisoners.
Now there is another way of viewing the Prigozhin affair.
During April of 2023, Prigozhin, and co-conspirators in the Russian Ministry of Defense, organized what appeared to be a serious dispute between them, although, as one might expect, it was never exactly clear what this dispute was about. This sowing of discord was to provide a plausible excuse to completely withdraw the Wagner group from Bakhmut (the town the Russians call Artyomovsk) as the Ukrainians were losing badly to the Wagner fighters.
On April 30 Prigozhin (the Wagner spokesperson, often incorrectly called the head of the group) accused the country's military leaders of "high treason" for not providing his group with ammunition, and threatened to withdraw Wagner from Bakhmut if it didn't receive more ammunition.
On May 5 Prigozhin stated that Wagner would pull out of Bakhmut on May 10. He stated this even though he had recently predicted that the (long delayed) Ukrainian counter-offensive would begin by 15 May. Thus, it is was somewhat likely, according to him, that the Ukrainian counter-offensive would occur after Wagner had been withdrawn, or as Wagner was being withdrawn. A Russian Patriot could hardly have missed that this might occur, and would not have planned a May 10 pullout. Yet Prigozhin does not even notice the problem. It is evidently not a concern of his.
In the evening of the next day the Ministry of Defense promised that the Wagner group would get all the arms they needed. Not having a sufficient excuse to pull Wagner fighters out, they stay, and fight, until May 20, when Wagner declared victory in Bakhmut. However, what was victory in Bakhmut? Victory in Bakhmut was simply the pushing of the Ukrainian army out of the contiguous suburbs of Bakhmut into the area west and south-west of the city. So, victory in Bakhmut, left intact most of the army that had been in Bakhmut, only now it was positioned west and south-west of the city, where much of it remains to this day. So even though we are told that the fight in Bakhmut is over, it actually still goes on.
On May 25 Wagner began their withdrawal from Bakhmut, and the transfer of positions to the Russian army. This was completed by June 1. So, the traitor Prigozhin had been able to arrange for Wagner fighters to be completely withdrawn and replaced by supposedly less capable forces. Not only that, but the Wagner forces left no fortified lines. These had to be cobbled together by the replacement forces and held against the entire Ukrainian army that had withdrawn from Bakhmut, and was now west, and south-west, of the city. This they were able to do. See this short video by Alexander Mercouris. Wagner's withdrawal let the pressure off the remaining Ukrainian forces giving them time to stabilize their defenses and plan future offenses. This is a similar strategy to that used by the traitor Surovikin at Kherson, and east of Kharkov. In one case you win the battle for the enemy by ordering your own troops to withdraw. In the other, you have the enemy escape defeat, by ordering your own troops to withdraw.
Bakhmut demonstrated that the Wagner forces are the best assault forces in the world. The Ukrainians (Jews hold all positions of power) know this and have worked to neutralize them. This is what the Prigozhin protest/rebellion was all about. Prigozhin called it a protest (against poor/treacherous treatment by the Ministry of Defense), but western "observers" called it a mutiny and worked to turn it into a fully fledged rebellion. That it was not a mutiny is obvious. The so called "march on Moscow" (from Rostov-on-Don to Moscow) was a march of over 1,000 kms, so they had no chance of getting to Moscow, let alone fighting there. The plan of the Russian traitors was to use the protest/rebellion as an excuse to expel Wagner forces from Ukraine. This is what happened. Many ended up in Belarus. The coup in Niger is part of the same operation, by the same Russian traitors. The plan is to have what is left of Wagner sent to Africa. Wagner is to be permanently removed from the fighting in Ukraine. They are to be sent somewhere else, anywhere else, anywhere other than back to Ukraine.
So, where in the past have we seen the withdrawal of an army just after they had won the battle, so that they ended up losing. Yes, when Hitler ordered Manstein to withdraw just after he had broken through the southern front, and essentially won the world war two battle of Kursk. About that battle, Field Marshal Erich von Manstein, the commander of Army Group South said;
"Speaking for my own Army Group, I pointed out (to Hitler) that the battle was now at its culminating point, and that to break it off at this moment would be tantamount to throwing a victory away."
It truly seems that the mad Jews are trying to replay world war two.
And, back to Tucker Carlson's interview with Todd Wood.
"We know (about the bioweapons) because Bob Kagan's wife, Victoria Nuland (U.S. Under Secretary of State for Political Affairs), who's not very bright, that's the one good thing you can say about her,... she's dumb enough to say it out loud,... announced in the (March 8) senate hearing; Oh by the way we have biolabs in Ukraine. And it turns out we do,... but why?" ... "I have my suspicions,... what their intentions are you can guess,... we've had the Covid-19 pandemic,..."
Really? Covid-19 is obviously a genetically engineered virus, but was it created in Ukraine?
Tucker Carlson has a whole episode on the Ukrainian crisis and another on the bio-labs. Here, 114M and here 22M.
This 2014 video reports on the U.S. support of Ukraine neo-Nazis and their role in the coup. 37M.
And, back to Covid.
What a top idea. Peter McCullough, and friends, are creating an alternative medical establishment. For this, and more, see the interview of Peter McCullough with Alex Jones. 133M
Peter McCullough teams up with crime writer John Leake to write the book; The courage-to-face-covid-19. This video is an introduction to their book about the criminal enterprise that was Covid-19. 67M
Peter McCullough is interviewed by Charlie Kirk. Here. 32M
Jeff Hays' documentary film "The Real Anthony Fauci" based on Robert F. Kennedy Jr.'s best-selling book of the same name. A story of massive corruption in the U.S. pharmaceutical industry.
The Dec 2022 vaccine accountability roundtable. Florida Governor Ron DeSantis asks the Florida Supreme Court to investigate "any and all wrongdoing in Florida with respect to Covid-19 vaccines." Here. 160M.
Senator Ron Johnson conducts a forum titled, "Covid-19 Vaccines: What They Are, How They Work, and Possible Causes of Injuries." Speakers Include Dr. Peter McCullough, Dr. Pierre Kory, Dr. Paul Marik, Dr. Robert Malone, ICAN Attorney, Aaron Siri, Esq., OpenVAERS Founder, Liz Willner, Edward Dowd, Dr. Harvey Risch, Dr. Ryan Cole, Journalist, Del Bigtree, and more. Full coverage, 640x360 229M; 1920x1080 1.4G. Highlights, 640x360 13M; 1920x1080 81M.
The Covid vaccinations seem to be responsible for a massive surge in cardiac problems among young fit people. I can't recall where I found this video, about it, but here is a web-page; 1884 Athlete Cardiac Arrests, 1310 of them Dead, after Covid vaccinations.
The 2022 Global Covid Summit. We declare that Pfizer, Moderna, BioNTech, Janssen, Astra Zeneca, and their enablers, withheld and willfully omitted safety and effectiveness information from patients and physicians, and should be immediately indicted for fraud. We declare government and medical agencies must be held accountable. Here. 78M.
Senator Ron Johnson, Dr. Pierre Kory, Dr. Robert Malone, and Dr. Peter McCullough discuss the Covid Cartel, Crony Capitalism, Big Pharma, and Vaccines. Here. 122M (August 2022)
Bret Weinstein talks to Dr. Pierre Kory about Ivermectin and the criminal campaign against it. An excellent video. 189M
CDC (Centers for Disease Control and Prevention) accused of fraud. Here. 41M. The New York Times article.
Dr Robert Malone calls for imprisonment of the guilty. Here. 31M.
Tucker Carlson interviews Dr Robert Malone. Here. 89M.
Del Bigtree interviews Dr Peter McCullough. Here. 150M.
Vaxxed the movie by Del Bigtree (part of The Highwire episode 259). Here. 158M.
Dr Robert Malone presents "An Overview of mRNA Vaccine Technology." Here. 60M.
Interview with Dr Meryl Nass. Was the Covid-19 virus genetically engineered? Here. 64M.
Dr Merritt Lee's talk about VAERS (Vaccine Adverse Event Reporting System) at the White Coat Summit. Here. 23M.
Dr Ryan Cole; why are there no autopsies for vaccine deaths. Here. 23M.
Roundtable discussion with Charlie Kirk, Dr Pierre Kory, Dr Ryan Cole, and Dr Flavio Cadegiani. Here. 143M.
The curtain close on covid theater, hosted by Florida Governor Ron DeSantis. Here. 109M.
Governor Ron DeSantis' fight to make monoclonal antibodies, and other treatments, available in Florida. Here 68M here 5M, and here 60M.
This video 168M, and the accompanying pdf 30M, from Dr. Richard Fleming, are very good. Unfortunately, in the past I have ignored him, as I was told (incorrectly) he was pushing the "graphene oxide in vaccines" garbage.
Ryan Cole, Robert Malone, and Denise Sibley give testimony for Tennessee House Bill 1871, March 9, 2022. Here. 52M.
In the video, Dr Ryan Cole states; Those who have had the vaccine don't have that ability (to clear the virus), because they don't have these little IgA secretory mops in their tears nose and throat and that's why we have seen a lot of those who have gotten, one, two, three shots still get covid, because they don't develop a full broad immunity like a natural infection does. Cole is wrong here. If it was a lack of IgA antibodies that allowed the vaccinated to be infected (with covid-19), then the vaccinated would only be infected at the same rate as the un-vaccinated. However, the vaccinated are now being infected at more than triple the rate of the un-vaccinated. This calls for some other explanation. See below.
Dr Ryan Cole also states; This virus has intermediate hosts, meaning the virus can bounce back and forth between animals and humans, and animals and humans, so unless you stick a shot in every bat and every pangolin and every deer, white tailed deer like to house this virus in their nasal mucosa as well. There are too many animal reservoirs for this kind of virus. Cole is completely wrong here. Thorough investigation has not found any animal reservoir where the virus bounces back and forth between animals and humans.
Another must watch video:
Dr. Pierre Kory talks about the war on hydroxychloroquine, ivermectin, and other cheap drugs that treat covid-19. "It's not about ivermectin. It's about the pharmaceutical industry capture of our agencies, and how our policies are all directed at suppressing and avoiding the use of cheap, repurposed drugs" he says. Download here. 73M. Bigger picture here. January 29, 2022.
Here is the Jan 24 2022 Covid-19: A Second Opinion hosted by Senator Ron Johnson at the U.S. Senate. 640x360, 379MB. A group of world renowned doctors and medical experts provide a different perspective on the global pandemic response, the current state of knowledge of early and hospital treatment, vaccine efficacy and safety, what went right, what went wrong, what should be done now, and what needs to be addressed long term. I will leave this video up for a few weeks. A larger version can be watched here and downloaded from here.
Dr Paul Marik makes the case for Ivermectin in this video. 39M. Some excerpts: After penicillin, Ivermectin is the second most important drug ever produced. It has saved hundreds of thousands of lives across this planet... It is on the WHO's list of essential medicines... 3.7 Billion doses have been dispensed to humans, to human beings, not horses. This is a remarkable drug. It has broad spectrum anti-parasitic, effective against a whole bunch of parasites, and... it is very effective against RNA viruses: HIV virus, zika virus, influenza virus, SARS-CoV-2 virus. In addition, what makes this a truly astonishing drug,... is it is a potent anti-inflamatory drug. And if anybody knows about Covid, Covid goes through stages. A viral replicative phase to a profound inflammatory phase. That's why Ivermectin is unique (it is not unique, hydroxychloroquine has similar properties) in that it treats across the spectrum (i.e., at all stages) of SARS-CoV-2. It's safe and well tolerated... You'll see an 83% improvement in prophylaxis trials, 66% improvement in early trials, 34% improvement in late trials, a 52% reduction in mortality. It reduces mortality by half.
Here are two must watch videos:
Dr Peter McCullough on vaccine injuries, vaccine deaths and omicron. Download here. 129M. Bigger picture here.
Dr Peter McCullough on the suppression of hydroxychloroquine, ivermectin, and other covid 19 treatments. Download here. 132M. Bigger picture here.
You can download the Dec 31, 2021, Joe Rogan, Dr. Robert Malone, interview from here. 156 MB. In the video, Malone states that the Government of India did not disclose what was in the covid kits distributed throughout Uttar Pradesh. Be that as it may, the Indian national newspaper the Hindu explicitly stated, on May 12, 2021, that kits containing ivermectin were to be distributed throughout the states Goa and Uttarakhand.
You can download the Jan 6, 2022, Pierre Kory, Greg Hunter interview from here. 66M. Pierre Kory concludes that the regulatory agencies (NIH, CDC, NIAID, etc) have been captured. By the way, the distorted video, and background noise, etc, is mostly added by the evil people who work for the video distributors. In some cases it is actually (deliberately) added by those who produce the video.
Here is Pierre Kory on FOX news (Jan 9, 2022). 11M. He warns of the capture of American agencies.
There are many more videos below.
The conspiracy against chloroquine (and hydroxychloroquine).
Chloroquine and SARS.
Chloroquine is a potent inhibitor of SARS-CoV infection, and spread, in vitro.
We report, that chloroquine has strong antiviral effects on SARS-CoV infection of primate cells. These inhibitory effects are observed when the cells are treated with (clinically admissible concentrations of) the drug either before or after exposure to the virus, suggesting both prophylactic and therapeutic advantage.[51]
We report on chloroquine,.. as an effective inhibitor of the replication of the severe acute respiratory syndrome coronavirus (SARS-CoV) in vitro. Chloroquine is a clinically approved drug effective against malaria. We tested chloroquine phosphate for its antiviral potential against SARS-CoV-induced cytopathicity in Vero E6 cell culture... The IC50 of chloroquine for inhibition of SARS-CoV (8.8 ± 1.2 µM) in vitro approximates the plasma concentrations of chloroquine reached during treatment of acute malaria... Chloroquine, may be considered for immediate use in the prevention and treatment of SARS-CoV infections.[52]
In vitro, is Latin for "in glass." It describes experiments, on cells, that have been grown outside of a living organism, e.g., cells that are grown in a test tube or petri dish. In vivo, is Latin for "within the living." It describes experiments, on cells, that are part of a whole living organism, such as a person, animal, or plant. The mentioned IC50 is the amount of chloroquine needed to inhibit SARS-CoV virus reproduction by 50%.
Chloroquine prevents terminal glycosyaltion of the ACE2 receptor.
These data provide evidence that ACE2 undergoes terminal glycosylation and that chloroquine at anti-SARS-CoV concentrations (1-10 µM) abrogates the process.[51]
Glycosyaltion refers to the attachment of sugar units/chains to a protein (like ACE2).
Previous studies of chloroquine have demonstrated that it has multiple effects on mammalian cells in addition to the elevation of endosomal pH, including the prevention of terminal glycosyaltion of immunoglobulins.[51]
Other mechanisms of action of chloroquine (antimalarials) include:
(1) interference with lysosomal acidification and inhibition of proteolysis, chemotaxis, phagocytosis, and antigen presentation;
(2) decreasing macrophage-mediated cytokine production, especially interleukin-1 and interleukin-6;
(3) inhibition of phospholipase A2 and thereby antagonizing the effects of prostaglandins;
(4) absorption and blocking of UV light cutaneous reactions;
(5) binding and stabilizing DNA;
(6) inhibition of T and B-cell receptors calcium signaling;
(7) inhibition of matrix metalloproteinases, and
(8) the inhibition of toll-like receptor signaling.[53]
Pre-treatment of cells with chloroquine leads to under-glycosylated ACE2 on the cell's surface. The SARS-CoV spike protein binds poorly to under-glycosylated ACE2 preventing the virus from entering, i.e., infecting, cells. (Remember, that the SARS-CoV virus spike protein attaches to the ACE2 receptor, and uses it to gain entry to the cell.) It takes 20-24 hours for the under-glycosylated ACE2 to replace the fully-glycosylated ACE2. Any shorter period, and there may remain sufficient fully-glycosylated ACE2 to allow infection of the cell.
Pretreatment with 0.1, 1, and 10 µM chloroquine reduced infectivity (of SARS-CoV) by 28%, 53%, and 100%, respectively.[51]
At correct doses, etc, chloroquine is known to be relatively safe.
However, like Tylenol (also known as Paracetamol), and like vitamin A, chloroquine is toxic if administered in high doses. A derivate, hydroxychloroquine, has similar properties to chloroquine but has been determined to be safer.
The inhibitory effects observed on SARS-CoV infectivity and cell spread occurred in the presence of 1-10 µM chloroquine, which are plasma concentrations achievable during the prophylaxis and treatment of malaria (varying from 1.6-12.5 µM) and hence are well tolerated by patients.[51]
It is known that chloroquine is effective against various autoimmune diseases, and thus may be effective in the later stages of SARS-CoV infections.
In recent years, antimalarials (like chloroquine) were shown to have various immunomodulatory effects, and currently have an established role in the management of rheumatic diseases, such as systemic lupus erythematosus and rheumatoid arthritis, skin diseases, and in the treatment of chronic Q fever. Lately, additional metabolic, cardiovascular, antithrombotic, and antineoplastic effects of antimalarials were shown.[53]
Summary: Chloroquine showed incredible promise against SARS-CoV.
With chloroquine showing such promise, the pharmaceutical industry felt it important to discourage further research. Thus, they published this paper [54] that concludes, incorrectly, that while chloroquine is active against SARS-CoV, in vitro, it is not active, in vivo, and therefore no further research, or trials, need be done.
In the study we find that chloroquine was delivered to BALB/c mice for 3 days, beginning 4 hours prior to virus exposure. It was observed that this did not inhibit the spread of the virus. From this it was concluded that chloroquine is inactive, against SARS-CoV, in vivo. This conclusion is clearly unwarranted as the authors gave only 4 hours, rather than the stated 20-24 hours, for the replacement of fully-glycosylated ACE2 by under-glycosylated ACE2 on the cell surface. Thus, enough fully-glycosylated ACE2 receptors remained to allow infection by SARS-CoV. This "oversight" is evidence that this paper is a hit-piece against chloroquine. Another telling sentence is;
The chloroquines, of which amodiaquin is a derivative, also inhibited virus replication at similar concentrations, although all were much more toxic than has been previously reported.[54]
Without any supporting evidence the authors claim that chloroquine (and the chloroquines) are much more toxic than previously reported. By the way, the previously reported levels, 1-10 µM, are extremely safe, and dosages of 100 times this amount are regularly given to sufferers of rheumatoid arthritis, and lupus. The sentence in question is simply a bald-faced lie, hence its vagueness. Here are some further quotes;
Patients with autoimmune diseases are given 250 mg/day of chloroquine (400 mg/day of hydroxychloroquine) regardless of height or weight of the patient. That is, cumulative doses greater than 100 times that used for prophylaxis or treatment of malaria.[55]
Chloroquine has been widely used to treat human diseases, such as malaria, amoebiosis, HIV, and autoimmune diseases, without significant detrimental side effects.[51]
The authors also bizarrely claim that chloroquine is active in vitro, and is also not active in vitro;
Chloroquine has already been approved for therapeutic use for several diseases. Because other groups had shown in vitro efficacy, it warranted further evaluation in the mouse SARS-CoV replication model even though in the present study the material was not found to be active in vitro (Table 1).[54]
Amodiaquin, a derivative of chloroquine was a rather potent inhibitor of SARS-CoV replication in cell culture in the present study (Table 1) and was evaluated in parallel studies with chloroquine.[54]
Looking at Table 1 we find;
Chloroquine IC50 = 2.5 ±0.7 µM
Amodiaquin IC50 = 2.5 ±0.7 µM
What does chloroquine having an IC50 value of 2.5 ±0.7 µM mean? It means that chloroquine at a concentration of 2.5 µM inhibits the replication of the virus by 50%. That is, chloroquine is active, in vitro. What does chloroquine having the same IC50 value as amodiaquin, mean? It means that chloroquine, as well as amodiaquin, is a potent inhibitor of SARS-CoV replication, in vitro. It is amazing that the authors are able to contradict themselves within the same paragraph. Further evidence that this is a hit-piece, is that this paper goes on to be widely quoted as proof that chloroquine is not active against SARS-CoV, in vivo, yet not one of those quoting the paper notice it's obvious flaws.
How has this paper effected Covid-19 research?
Note that the Covid-19 virus is called SARS-CoV-2 because it is very similar to SARS-CoV (76% amino acid identity). The main differences being the added furin cleavage site, and the changed RBD (receptor binding domain). Both changes make the virus more contagious to humans. Although more contagious, SARS-CoV-2 is less dangerous. SARS-CoV is now often called SARS-CoV-1.
ACE2 is also the receptor used by SARS-CoV-2 (the Covid-19 virus) to infect cells. It is known that fully-glycosylated ACE2 is a very good fit to the SARS-CoV-2 receptor binding domain.[56] Thus, it is almost certain that ACE2 without terminal glycosylation will bind poorly, and chloroquine will be an effective prophylactic for Covid-19.
However, because of this hit-piece, any promise of chloroquine against SARS-CoV-2 that is shown, in vitro, will be accompanied by doubt about it, in vivo, and the necessary trials will likely not be done. As it turns out, chloroquine did show great promise against SARS-CoV-2, in vitro, and numerous clinical trials were initiated around the world. However, the pharmaceutical industry managed to halt most of these trials. For example, in order to bad-mouth chloroquine, the Brazilian Borba trial [57] gave near fatal doses of chloroquine to very sick patients. This had the desired effect of killing some of them, and making chloroquine appear quite dangerous. Criminal charges [58] were bought against the trial administrators.
Chloroquine and HIV/AIDS.
By 1987 researchers had found that chloroquine was active against HIV, and it appeared likely that chloroquine would eventually replace the very expensive, very toxic, drug AZT (azidothymidine, also known as zidovudine and retrovir) used in the treatment of AIDS, from which Burroughs Wellcome (GlaxoSmith-Kline) was making a killing. At US$10,000 a year for AZT the drug company stood to lose a very profitable product.
Some introductory remarks about HIV.
During the production of new viral particles HIV causes the cellular machinery to produce (among other things) the molecule gp160. This is then cleaved (at a furin cleavage site) by cellular proteases (enzymes which cut proteins) of the furin family, to produce gp120 (which contains the receptor binding domain of the HIV virus) and gp41 (which is the HIV fusion protein). These two remain associated through non-covalent interactions, and end up, as a unit called the gp120-gp41 complex, embedded in the viral membrane. The gp120 molecule sits in the fluid surrounding the virus loosely attached to one end of the gp41 molecule, the other end of which is embedded in the viral membrane. The gp120-gp41 complex is functionally equivalent to the covid virus spike protein. See [1] and [2]. The notation gp160 indicates a glycoprotein (a protein with multiple sugar units/chains attached to it) with a molecular weight of about 160 kilodaltons. Similarly, for gp120 & gp41.
To reproduce the HIV virus needs to get into a cell and use the cell's chemical machinery. To do this the gp120 part of the gp120-gp41 complex binds to a particular molecule, called CD4, found embedded in the membrane (surface) of certain human immune cells. Once bound, a complex series of changes, which involves the shedding (release) of gp120, enables the gp41 part of the complex to harpoon the cell membrane, pull the viral and cell membranes together, and fuse them. This has the effect of emptying the viral RNA into the cell cytoplasm where it hijacks the cellular machinery to produce more virus.
"Like all other viruses, HIV cannot reproduce on its own, requiring the reproductive apparatus of a cell that the virion “infects.” HIV gains entrance to a target cell mainly through a glycoprotein (gp120) of the viral envelope protein. Since HIV is a retrovirus (re = reverse, tr = transcriptase), it contains the viral enzyme RNA-dependent DNA polymerase (reverse transcriptase) that transcribes DNA from the virion's two identical strands of RNA containing all of virion's genetic information. The double-stranded DNA made by the virion's reverse transcriptase migrates into the nucleus of a target cell and is inserted with the aid of another viral enzyme (integrase) into the cell's genome. Once the virion's DNA is integrated into the cell's reproductive apparatus, exact copies of the virus are produced when the infected cell is stimulated to replicate. The proviral DNA is transcribed into viral RNA, and messenger RNA (mRNA) and new virions are synthesized which can infect new cells. Since zidovudine inhibits the HIV reverse transcriptase enzyme and stops proviral DNA chain synthesis in vitro, the drug held out promise of being effective in slowing or stopping the progression of the infection in vivo."[3]
In AIDS, the HIV virus (variously designated LAV, HTLV-III, or ARV) debilitates the body's immunological defenses, by infecting and killing CD4+ lymphocytes (CD4 expressing lymphocytes) and shedding immuno-suppressive gp120 from infected cells. CD4 lymphocytes defend against invading foreign matter; thus, their destruction renders the body susceptible to a spectrum of diseases. In addition to infecting CD4 lymphocytes, HIV also infects other immune system tissue such as monocytes, nervous system tissue, intestinal tissue and some bone marrow cells. Monocytes and macrophages (matured monocytes) are not killed outright by HIV. Rather, it is believed that, once infected, these cells serve to continuously incubate the virus.
The historical record.
In Nov. 1986, Maddon et al., published the paper [4] where they observed that; "If JM cells are exposed to ammonium chloride (for 6 hr) either at the time of addition of (the HIV) virus or within 30 min after the addition of virus, we observe greater than 95% inhibition of viral infection."
This paper was big news among AIDS researchers as it finally established that the HIV cellular receptor is CD4 (once called T4). Everyone in the field would have known of this paper. The paper also reported that ammonium chloride (NH4Cl) inhibited infection by more than 95%. Yet, somehow, this equally important result never made the headlines.
Maddon, and his team, explain that; "Previous studies have described two distinct pathways of entry for enveloped viruses. Some viruses fuse directly with the plasma membrane, releasing their nucleocapsids into the cytoplasm, whereas others are internalized by receptor-mediated endocytosis. The acidic environment of the endosome then facilitates fusion of the viral envelope with the limiting membrane of the vacuole. Infection by viruses that enter cells via the endocytic pathway can be inhibited by treating cells with agents such as weak bases which deacidify the endosome (Helenius et al., 1980,...)."
Checking the references (at the end of their paper) one finds that chloroquine, tributylamine, amantadine, methylamine and ammonium chloride (NH4Cl) are among the weak bases referred to. The reference [5] (Helenius et al.) presents the following table from which one can conclude that chloroquine is the most effective of these agents when dealing with the Semliki Forest Virus and thus potentially the most effective against HIV. They state: "For further studies, we used chloroquine as it was the most effective inhibitor."
Table 1: Effect of Lysosomotropic Agents on SFV Infection
Inhibitor PFU/ml Cells
Inhibitor concentration x 10-6 infected
mM
None - 140.0 64
Tributylamine 1.0 66.0 31
Amantadine 0.5 14.0 19
Methylamine 10.0 3.6 10
Chloroquine 0.1 1.2 <1
NH4Cl 10.0 0.8 <1
Given this, why did Maddon not report results for chloroquine. Given that chloroquine would have been expected to be the most effective against AIDS, even a negative result would have been of interest. We now know, for example, that 12.5 μM/L chloroquine inhibits HIV infection by 90%. Maddon, and team, would have certainly investigated chloroquine and thus could have reported that chloroquine, at the clinically achievable concentration of 12.5 μM/L, inhibited HIV infection by 90%, but they chose to report that ammonium chloride, at the very toxic concentration of 20,000 μM/L, inhibits HIV infection.
So, as you can see, the conspiracy against chloroquine was already up, and running, in 1986.
Another reason Maddon, and team, should have reported on the effect of chloroquine, is that, five months before their paper, Thorens, and Vassalli, had reported that both chloroquine, and ammonium chloride, prevented the terminal glycosylation of immunoglobulins (and thus chloroquine, as well as ammonium chloride, should inhibit HIV infection by preventing the glycosylation of the gp120-gp41 complex).[6]
In Feb. 1987, Bruce Kagan wrote a letter to the editor of The Western Journal of Medicine, pointing out the potential of chloroquine to combat HIV.[7] Although published four months after Maddon et al's article appeared (in the journal Cell) Kagan only references a research announcement to Maddon's work (in the journal Science) that did not report the fact that ammonium chloride greatly inhibits HIV infection. Adding this fact would have significantly strengthened Kagan's case. Importantly, Kagan notes that, even if the concentration of chloroquine needed in vitro may have been severely toxic, chloroquine becomes highly concentrated in certain tissues in vivo, notably in the cerebrospinal fluid (10 to 30 times plasma level), the reticuloendothelial system (6,000 times), and leukocytes (100 to 200 times), meaning that in certain situations the use of very high concentrations of chloroquine in vitro is justified, as the concentration in vivo (in the plasma) remains safe.
From 1986 to 1995 the conspirators dissuaded all from conducting the clinical trials called for by Kagan.
Maddon, and team, further say; "These results (the inhibition of viral infection) are consistent with a mechanism of viral entry which involves endocytosis of the CD4-AIDS virus complex and low pH-induced fusion of the viral envelope with the limiting membrane of the endosome, releasing the viral nucleocapsid into the cytoplasm of the cell. It should be noted, however, that ammonium chloride and amantadine may exert other inhibitory effects on virus production and additional experiments are required before we may conclude with certainty that the primary route of entry of the AIDS virus is via receptor-mediated endocytosis."
Later, the speculation that HIV enters cells via receptor-mediated endocytosis was proved false. Rather, it was found that ammonium chloride caused the viral progeny to be non-infectious. This was verified in another paper which also avoids reporting on chloroquine. We find that; "Maddon et al. (1986), however, suggested (reported) that NH4Cl inhibits HIV-1 replication. A possible explanation for this discrepancy is our finding that weak bases inhibit the release of infectious HIV-1," and "Our experiments indicate that the budding and release of HIV-1 virions is not itself affected by NH4Cl treatment, because there is no significant reduction of particles containing RT (reverse transcriptase), but that the virions released in the presence of NH4Cl have reduced infectivity."[8]
What we learn is that chloroquine does not stop the entry of (fully functional) HIV into cells. What chloroquine does is it causes the production of non-functional viral offspring which are non-infectious, i.e., incapable of infecting further cells. Chloroquine does not directly reduce the number of new virions produced. It reduces the number of functional virions produced, which reduces the number of newly infected cells. So, chloroquine indirectly inhibits viral entry into cells and thus inhibits replication of the virus.
In another paper, which also strangely avoids reporting on chloroquine, we find that; "An unexpected result from our studies was the marked sensitivity of gp160 cleavage to NH4Cl. A recent report [8] has shown that NH4Cl treatment of HIV-1-producing T cell and monocyte cell lines results in a major loss (>95%) in titer of infectious virions. Coupled with our findings, these results suggest that, in the presence of NH4Cl, noninfectious nucleocapsids that lack gp120 are released into the medium and point to the intracellular cleavage of gp160 as a requirement for infectivity."[9]
Actually, they did mention chloroquine, but tell you nothing; "Partial inhibition was also observed with the weak base chloroquine (data not shown)."
As mentioned above gp160 is cleaved to produce gp120 and gp41. These last two remain non-covalently associated, and are added, as a unit, to HIV virions during assembly. Some say that uncleaved gp160 is directed to a lysosome and broken down, others, that it is added to virions, or perhaps both occur. Whatever the case, virions produced in the presence of NH4Cl are non-infectious.
So, what was the response of the scientific community to the news that various weak bases inhibited HIV infection by greater than 95%. Did they yell from the roof-tops that a cure for AIDS may be at hand? Did hundreds of scientists enter the area to confirm, and extend. Did they immediately set up clinical trials to check that in vitro results held in vivo? No, they did none of that, however they did hurry out an entire issue of Scientific American (October 1988) singing praises of the very toxic drug AZT, and other retroviral drugs pushed by the pharmaceutical companies, without ever mentioning NH4Cl, or chloroquine, or any of the other aforementioned weak bases.[10]
Papers claiming that chloroquine is effective against HIV were published in the 5th International Conference on AIDS in June 1989.[11] Here are the abstracts of two;
Abstract: "The effect of chloroquine, an anti-malarial drug known to affect cellular exocytic pathways, was studied in two retroviral systems: human immunodeficiency virus (HIV-1) and avian reticuloendotheliosis virus (REVA). With chloroquine treatment of virus infected cells, significant size reduction of the cell and virus associated surface glycoproteins, gp90 of REV-A and gp120 of HIV-1, was observed. In the case of HIV-1, extracellular virions derived from treated cells contained very little gp120. Infectivity and reverse transcriptase assays carried out with HIV-1 demonstrated that by chloroquine treatment virus yield was reduced and noninfectious virions were released. The data suggests that inhibition by chloroquine is most likely due to interference with terminal glycosylation in the trans-Golgi network. Studies are in progress to determine the effect of chloroquine on HIV-1 and its relatives produced by other cell types e.g. monocytes/macrophages and to evaluate whether chloroquine and its existing analogs or newly synthesized related weak bases could be useful either alone or in combination with other drugs to attack various stages of the virus life cycle."[12]
It is interesting that chloroquine showed great promise against both HIV and REV-A, and that studies were in progress to extend the reported observations. None of these studies were ever published, in fact, the sentence "Studies are in progress...." was dropped from the journal version of the article.[13]
Abstract: "The treatment of HIV infected monocytes by chloroquine resulted in the formation of intracellular vacuoles containing virions in various stages of degredation similar to those observed in SFV (Semliki Forest Virus) infected cells treated with lysosomotropic agents (1) and in HSV (Herpes Simplex Virus) infected cells expressing a glycoprotein that interferes with the fusion reaction of virion envelope with the cell-membrane (2). By contrast such intravacuolar accumulations of virions were not observed in HIV infected monocytes not treated by the drug."[14]
This study was never published in a journal so it would be of great interest to find notes from the talk. Once again chloroquine is reported to show great promise against HIV. In this case against the infection of monocytes. Any action against the infection of monocytes is of particular interest as drugs like AZT and DDC are essentially useless in this regard. The reason for this is:
"Before a nucleoside analogue (like AZT and DDC) can be incorporated into a strand of nucleic acid it must be phosphoryiated by a kinase (converted to a nucleotide). T-cells which are capable of rapid division possess high levels of kinases to phosphorylate antiviral drugs like AZT and DDC. Monocytes which divide only under special circumstances, possess relatively low levels of the requisite kinases and the nucleoside derivatives do not protect them against infection. The simple solution would be to administer the phosphoryiated form of the nucleotide analogues. These, however, are not transported across cell membranes. Thus the current nucleoside analogue therapy is ineffective in protecting monocytes from HIV infection."[15]
In 1993, Sperber et al reported that: "Hydroxychloroquine inhibited HIV-1 replication (>75%), as measured by reverse transcriptase activity, in the primary T cells and monocytes as well as the T cell (CEM) and monocytic cell lines (U-937). Hydroxychloroquine itself had no anti-reverse transcriptase activity and was not toxic to the cells at concentrations inhibitory to viral replication."[16]
Although Sperber et al claim a greater than 75% inhibition of replication in the paper's abstract, there is no further reference to this claim in the paper. Examining the graphs in figures 1 & 3 we find that 100 μM/L hydroxychloroquine inhibits reverse transcriptase activity by 68% in primary T cells, by almost 75% in primary monocytes, by 30% in CEM cell lines and by 81% in U-937 cell lines.
Reverse transcriptase activity measures the overall viral yield. Thus it measures both infectious and non-infectious virus. The fact that less than one in 10,000 HIV virions are infectious[17] means that the inhibition of replication is less than the inhibition of infectivity. Thus the inhibition of infectivity is a much better choice (than the chosen reverse transcriptase activity) with which to measure the effectiveness of hydroxychloroquine. From Tsai, et al, (one of their references) we read;
"From our studies, chloroquine appeared to be an effective inhibitor of HIV-1 by reducing both the yield (measured by reverse transcriptase assay) and infectivity (measured by a microtiter syncytial-forming assay with cloned CEM cells) of the virus produced in chronically infected cells. This effect of chloroquine is consistent with the reported inhibition of HIV infectivity by NH4Cl." and "The reduced RT (reverse transcriptase) activity is likely due to the decrease in the number of virions produced. However, the extent of reduction of RT was apparently less than that of infectivity (see Table 1)."[13]
We fit the relevant curves to Tsai et al's data to obtain the following graph;
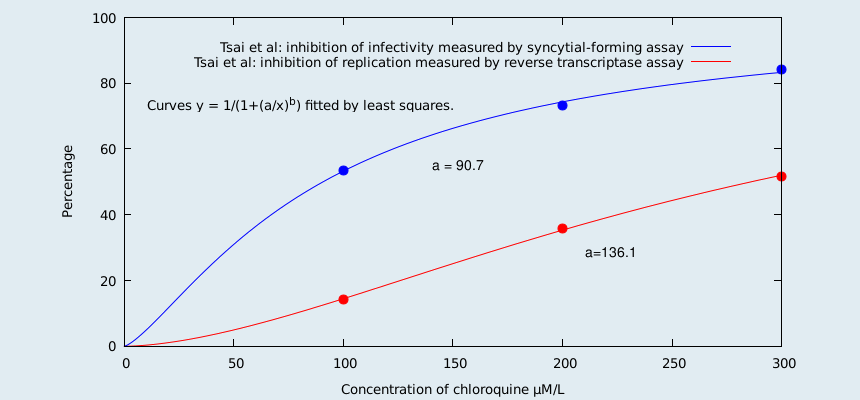
For chloroquine (as can be seen from the graph) the inhibition of replication is much less than the inhibition of infectivity. Since chloroquine and hydroxychloroquine are chemically similar, and are acting as lysosomotropic agents, one expects this to also be true for hydroxychloroquine. In a short paragraph (split over 3 pages) at the end of their paper, Sperber et al do actually measure the inhibition of infectivity. They report that the production of infectious virus was inhibited by 77% in CEM cells, and 80% in U-937 cells. They do not report the concentration of hydroxychloroquine used, but we infer that there is a concentration where the inhibition is as stated. So, there you have it. Sperber et al measured the inhibition of infectivity, and also the inhibition of replication, and then choose the method that gave the worst results. Yes, looks like this is yet another paper written in aid of the war against cheap drugs.
And then, there are the lies of omission. They say, not a word, about chronically infected cell lines. Why is this? Given how important the topic was in AIDS research it is a rather glaring omission. The reason is simple, AZT has little, or no effect, on the production of infectious virus by chronically infected cells, whereas, hydroxychloroquine is effective against such. One must never be told that hydroxychloroquine is a much better drug than AZT.
"Zidovudine (AZT) and similar RT inhibitors have little or no effect on the production of infectious virions by chronically infected cells."[18]
That Sperber et al are funded by Sanofi-Winthrop Pharmaceuticals, comes as no surprise.
Early on, the most up to date research seems to be not in scientific papers, but in patent applications. See [16] for what was known before Feb. 1988. A patent was filed on 14 September 1990, by Ellen Vitetta, and & Jonathan Uhr, for the Board of Regents of The University of Texas System. It states;
"It has been found by the inventors that chloroquine alone exhibits significant anticellular activity against HIV-infected cells. Furthermore, chloroquine will act in concert with the toxin conjugates of the present invention to improve their anti-HIV efficacy. In both instances, it is suggested or has been found that the amount of chloroquine to be administered to a patient in need of such therapy will be similar to that amount normally administered for other indications of chloroquine, such as in the treatment of malaria."[19]
Another patent [15 May 1991], by the same pair, was filed in May 1991 which claimed that chloroquine selectively killed HIV-infected cells [NIH grant AI-27336]. This should have been big news.
Abstract: "The present disclosure demonstrates the use of chloroquine to selectively kill HIV-infected cells. The invention involves, in part, the use of chloroquine in treatment regimens for the treatment of HIV-infected individuals, wherein chloroquine is administered to such individuals in pharmacologically effective doses. While it is proposed that chloroquine may be administered to HIV-infected individuals alone, it is believed that additional benefits may be realized through the administration of chloroquine in combination with other agents such as anti-HIV immunotoxins (e.g., CD4-based immunotoxins, or anti-gp4I or anti-gp120-based immunotoxins). In these embodiments, it is believed that chloroquine will act synergistically with the anti-HIV immunotoxins to effect a selective killing of HIV-infected cells."[20]
If, as reported in the patent, chloroquine selectively kills HIV in vitro this makes it "orders of magnitude" better than AZT. Also, due to the nature of the action of weak bases like chloroquine, this is likely true in vivo. As far as I am aware, the fact that chloroquine selectively kills HIV in vitro never makes it into the scientific journals. Not even as an experiment to investigate the possibility. The patent also states;
"In in vitro IC50 tests, HIV-infected human H9 T-cells have been found by the inventors to be up to fifty-fold more sensitive to chloroquine than uninfected cells. Studies also indicate that the concentrations necessary to kill infected cells in vitro are within the dosage range used for in vivo treatments of other diseases such as malaria." and "Chloroquine's quality of specifically concentrating in the very tissues likely to be infected by HIV means that chloroquine can be delivered to these tissues in an appropriate dosage while exposing the tissues unsusceptible to infection to a relatively low dosage. This quality of chloroquine should minimize the potential for adverse reactions in the unsusceptible tissues."
At the end of 1986 it was clear that clinical trials testing chloroquine (as requested by Bruce Kagan) needed to be done. Yet for nearly a decade no trial was done, In 1995 a small 8 week long clinical trial is finally done.
"In 1995, Sperber et al conducted a randomized, double-blinded, placebo-controlled study of 40 asymptomatic HIV-positive patients with CD4 counts between 200 and 500 cells/mm3. Half of the patients received HCQ, the other half served as controls. The placebo group showed an increase in viral RNA load (bad), no change in IL-6 levels, and a decline in both the CD4 percentage (bad) and T-cell proliferation to Candida (bad). In contrast, the HCQ-treated group experienced a decline in HIV-1 RNA (good) and IL-6 (good), and maintained CD4 percentages (good) and mitogen and antigen-specific T-cell proliferative responses (good)."[21]
This clinical trial was written up as "Hydroxychloroquine treatment of patients with Human Immunodeficiency Virus Type 1."[22] Is this another paper written to mislead? Looks like it. Here we have a clinical trial done at the Mount Sinai Medical Center in New York City, an epicenter of the AIDS crisis, with thousands, and thousands, of patients who would have been willing to participate in an eight week trial to test hydroxychloroquine. Yet only 42 participate. Clearly, the decision to proceed with an under-powered clinical trial must have been deliberate.
"Sperber extended his study by comparing HCQ to zidovudine (AZT) in the same group of patients (as in the 1995 clinical trial). There was no statistical difference in the magnitude of RNA viral load decline, levels of cultured virus, p24 antigen, or CD4 counts. IL-6 and serum IgG levels were significantly reduced (good) in the HCQ group, but not the zidovudine group."[21]
The extended study appeared in 1996 as "Inhibition of HIV-1 replication by Hydroxychloroquine: Mechanism of action and comparison with Zidovudine (AZT),"[23] This is yet another paper in the war against cheap drugs. Here are some quotes from the paper;
"The effective concentration of HCQ to reduce viral replication by 50% (ED50) was 0.01 mmol/L (10 μM/L) in the 63-cells and 0.1 mmol/L (100 μM/L) in the SP cells." and "Both HCQ and AZT (ZDV) had anti-HIV-1 activity in the pretreated, recently infected SP and 63 cells (Figure 4), although HCQ was less effective. The ED50 for AZT (ZDV) was 0.001 mmol/L (1 μM/L), while the ED50 for HCQ was 0.01 mmol/L (10 μM/L)."[23]
A quick look at Figure 4 tells you that some of the ED50 numbers must be wrong. In the hope that the graphs are less in error than the text, I have calculated the ED50 numbers from them. (One fits the data to a graph of the form y=100/(1+(a/x)^b) by least squares, which determines a = ED50 and b = the Hill coefficient.) I have made the assumption that ED50 = IC50, i.e., that the drugs completely inhibit the relevant biological activity at high dose. This gives somewhat different results:
ED50[AZT; newly infected 63 cells] = 0.3 μM/L (inferred to be 1 μM/L).
ED50[HCQ; newly infected 63 cells] = 0.4 μM/L (said to be 10 μM/L).
ED50[AZT; newly infected SP cells] = 0.1 μM/L (inferred to be 1 μM/L).
ED50[HCQ; newly infected SP cells] = 49.0 μM/L (said to be 100 μM/L).
ED50[HCQ; chronically infected 63HIV cells] = 9.1 μM/L (not stated).
ED50[AZT; chronically infected 63HIV cells] is undefined, as AZT has no effect at all.
ED50[HCQ; chronically infected SPH cells] = 39.0 μM/L (not stated).
ED50[AZT; chronically infected SPH cells] is undefined, as AZT has no effect at all.
"Either AZT or HCQ was maintained in culture at a concentration of 1 μmol/mL (1000 μM/L)."[23] One assumes that this is an error.
Note that cells were harvested daily for 14 days to further! assess the toxicity of HCQ. However, no such experiment was done to access the toxicity of AZT.
"We next compared the anti-HIV-1 effect of HCQ with zidovudine (AZT) in both newly and chronically HIV-1-infected T-cell and monocytic cell lines (63 and 63HIV). HCQ suppressed HIV-1 replication in a dose-dependent manner in both recently and chronically infected T-cell and monocytic cell lines. In contrast, zidovudine (AZT) pretreatment had potent anti-HIV-1 activity in the newly infected T and monocytic cells but not in chronically infected cells."[23]
The above is very deceptively worded. Let's add some details (in square brackets):
"We next compared the anti-HIV-1 effect of HCQ with zidovudine (AZT) in both newly and chronically HIV-1-infected T-cell [lines; i.e., SP and SPH cells] and monocytic cell lines (63 and 63HIV). HCQ suppressed HIV-1 replication in a dose-dependent manner in both recently and chronically infected T-cell [lines; SP cells; ED50 = 49 μM/L and SPH cells; ED50 = 39.0 μM/L] and monocytic cell lines [63 cells; ED50 = 0.4 μM/L and 63HIV cells; ED50 = 9.1 μM/L]. In contrast, zidovudine (AZT) pretreatment had potent anti-HIV-1 activity in the newly infected T [cells; SP cells; ED50 = 0.1 μM/L] and monocytic cells [63 cells; ED50 = 0.3 μM/L] but not in chronically infected cells [where AZT has zero effect on SPH cells and zero effect on 63HIV cells]."
Stating that AZT pretreatment had potent anti-HIV-1 activity in the newly infected T and monocytic cells but not in chronically infected cells, suggests that AZT has some value for chronically infected cells. However, AZT has no anti-HIV-1 activity, at all, in chronically infected cells.
"AZT has no effect on virus replication of an already integrated virus."[24]
"Although AZT prevented acute HIV infection of susceptible cells, it did not prevent the induction of HIV expression in the infected cell lines."[25]
"Compounds currently used for AIDS therapy, such as AZT, DDL and other nucleoside analogues, are inhibitory to HIV infection of T cells in vitro at low concentrations. They are, however, ineffective as anti-HIV agents when the virus is already integrated into the T-cell DNA, so that the cells are chronically or latently infected."[26]
Let's find out a little more about AZT.
"Current AIDS therapy which is directed towards protecting uninfected cells, consists of oral dosing about every four hours with nucleoside analogues (such as AZT and DDC) which inhibit viral RNA replication. Although these drugs inhibit viral replication at concentrations of 50-500 μM, at higher concentrations (~1000 μM) they also inhibit the DNA polymerase of healthy cells which is required for cell division. The current therapy requires very large doses of drugs (up to a gram/day). Because the drugs are taken orally and in a form that is absorbed by all cells, the entire body is exposed to them. Toxicity is a serious limitation to their use; anemia being one of the most severe side effects. Because nucleoside derivatives must be phosphoryiated before they can be incorporated into DNA (and express their chain disrupting activity) they require kinases which are not present in equal amounts in all cells susceptible to viral infection. Thus the oral nucleoside analogue therapy, which is ineffective against already infected cells, is only able to protect those susceptible cells which can convert high concentrations of nucleosides into nucleotides (i.e., dividing cells). For these reasons this therapy is limited and the progression of the disease is only slowed."[15]
Although very toxic in the quantities administered, AZT did show promise of being effective in slowing or stopping the progression of the infection in vivo. However as early as 1987 it was known that AZT only inhibits viral replication for a short period.
"Initially AZT works to inhibit the virus but after a period AZT treated cells produce as much virus as non-treated cells."[27]
"Probably, a majority of the DNA copies initiated by RT in newly infected cells are prematurely terminated, but if at least one complete copy is made (it only takes one), a cell may go on to produce progeny virus. Whether virus spread occurs by cell-free virus or by cell-to-cell contact, (even) cultures treated with 25 μM AZT (the highest dose tested) eventually produced as much virus as the non-drug-treated infected cultures. These results were confirmed by the detection of unintegrated viral DNA in drug-treated H9 cultures when they began producing virus at high levels. The unintegrated viral DNA in these drug-treated cultures was present in quantities similar to those in non-drug-treated infected cultures."[27]
"The inability of zidovudine (AZT) therapy to prevent infection after exposure to HIV is described in a report by Lange et al. In a tragic hospital accident, a seronegative 58-year-old heterosexual man was injected with HIV-contaminated blood. This mistake was realized within minutes of its occurrence, and within 45 minutes after exposure to HIV-1, massive zidovudine therapy was begun. In spite of the daily administration of large amounts of zidovudine (AZT), the patient seroconverted (started producing antibodies against HIV as a result of the HIV infection becoming established) on the 41st day after exposure. These results suggest that the HIV-1 infection could be established by cell-to-cell transmission of the virus or that zidovudine may not completely inhibit reverse transcription. The latter explanation could account for the limited ability of zidovudine therapy to stop HIV replication in vivo and extend the survivability of HIV infecteds with bureaucratic AIDS."[27]
It is important to note that AZT only inhibits replication. It does not, and cannot kill HIV. It's mode of action makes it clear that AZT cannot kill the virus.
In his book "Poison By Prescription. The AZT story," John Lauritsen states:
"In my articles (in this book) I have analyzed the studies that allegedly demonstrate AZT's effectiveness, and have concluded that there is no scientifically credible evidence that AZT has benefits of any kind. Documents which the Food and Drug Administration (FDA) was forced to release under the Freedom of Information Act revealed that the Phase II ("double-blind, placebo-controlled") AZT trials were worthless. The researchers covered up the fact that the study had become unblinded (thus violating the basic test design). Protocol violations were overlooked. Worst of all, the researchers deliberately used data that they knew were false. These fraudulent trials were the basis of government approval of the drug. Another study often cited as proof of AZT's benefits concerns patients who received AZT after the Phase II trials were prematurely terminated. I have written an extensive analysis of this study, which is a rotten mixture of incompetence and dishonesty. Through colossal incompetence the researchers lost track of 1120 patients, not knowing if they were even alive or dead. They then used statistical projection methods to guess what results they would have obtained if they had not lost the 1120 patients, presented their guesses as actual survival statistics, and made a number of grossly invalid comparisons in order to claim that AZT had extended lives. This "research" is a blatant exercise in disinformation, proving nothing except how farcical are the "peer review" standards of medical journals."[28]
A very similar article could be written about the Covid scam.
"AZT is the only antiretroviral drug that has received FDA approval for treatment of AIDS since the epidemic began ten years ago, and the decision to approve it was based on a single study that has long been declared invalid. The study was intended to be a “double-blind placebo-controlled study.” In such a study, neither patient nor doctor is supposed to know if the patient is getting the drug or a placebo. In the case of AZT, the study became unblinded on all sides, after just a few weeks. Both sides contributed to the unblinding. It became obvious to doctors who was getting what because AZT causes such severe side effects that AIDS per se does not. Furthermore, a routine blood count (watching for cytomegalovirus infections), which clearly shows who is on the drug and who is not, wasn't whited out in the reports. Both of these facts were accepted and confirmed by both the FDA and Burroughs Wellcome, who conducted the study. Many of the patients who were in the trial admitted that they had analyzed their capsules to find out whether they were getting the drug. If they weren't, some bought the drug on the underground market. Also, the pills were supposed to be indistinguishable by taste, but they were not. Although this was corrected early on, the damage was already done. There were also reports that patients were pooling pills out of solidarity to each other. The study was so severely flawed that its conclusions must be considered, by the most basic scientific standards, unproven. The most serious problem with the original study, however, is that it was never completed. Seventeen weeks into the study, when more patients had died in the placebo group, the study was stopped, five months prematurely, for “ethical” reasons: It was considered unethical to keep giving people a placebo when the drug might keep them alive longer. Because the study was stopped short, and all subjects were put on AZT, (and any treatment without AZT falsely declared unethical) no scientific study can now be conducted to prove unequivocally whether AZT does prolong life."[29]
From this, it truly looks like the AIDS crisis was used as a model for the Covid scam.
How did the extremely toxic drug AZT become the standard of care in the AIDS crisis, while chloroquine and hydroxychloroquine were side-lined?
How did the extremely toxic drug Remdesivir become the standard of care in the Covid scam, while chloroquine and hydroxychloroquine were side-lined?
[1] The HIV-1 envelope glycoproteins: folding, function and vaccine design.
[2] HIV Infection: The Cellular Picture. Scientific American, October 1988.
[3] The Impact of Zidovudine (AZT) Therapy on the Survivability of Those with the Progressive HIV Infection.
[4] The T4 (CD4) gene encodes the AIDS virus receptor and is expressed in the immune system and the brain.
[5] On entry of Semliki forest virus into BHK-21 cells.
[6] Chloroquine and ammonium chloride prevent terminal glycosylation of immunoglobulins in plasma cells without affecting secretion.
[7] Lysosomotropic Agents in AIDS Treatment.
[8] Human immunodeficiency virus infection of CD4-bearing cells occurs by a pH-independent mechanism.
[9] Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160.
[10] Scientific American, October 1988.
[11] Fifth International Conference on AIDS, Quebec, Canada June 4-9, 1989.
[12] Abstract. Session M.C.P.66. Inhibition of human immunodeficiency virus infectivity by chloroquine.
[13] Inhibition of Human Immunodeficiency Virus Infectivity by Chloroquine.
[14] Abstract. Session M.C.P.119. Effect of chloroquine on HIV-1 infection of monocytes.
[15] Methods and compositions for the use of hiv env polypeptides and antibodies thereto.
[16] Inhibition of Human Immunodeficiency Virus Type 1 replication by Hydroxychloroquine in T Cells and Monocytes.
[17] Factors underlying spontaneous inactivation and susceptibility to neutralization of Human Immunodeficiency Virus.
[18] Human immunodeficiency virus type 1 protease inhibitors irreversibly block infectivity of purified virions from chronically infected cells.
[19] Patent PCT/US90/05234. Therapeutic compositions; Methods for treatment of HIV infections.
[20] Patent PCT/US91/03383. Method for killing HIV-infected cells.
[21] Effect of chloroquine on human immunodeficiency virus (HIV) vertical transmission.
[22] Hydroxychloroquine treatment of patients with Human Immunodeficiency Virus Type 1.
[23] Inhibition of HIV-1 replication by hydroxychloroquine: Mechanism of action and comparison with zidovudine.
[24] Inhibition of Replication and Cytopathic Effect of Human T Cell Lymphotropic Virus Type III.... Virus by 3'-Azido-3'-Deoxythymidine In Vitro.
[25] Interferon-α but not AZT suppresses HIV expression in chronically infected cell lines.
[26] Inhibition of the production of HIV-1 from chronically infected H9 cells by metal compounds and their complexes with L-cysteine or N-acetyl-L-cysteine.
[27] Resumption of virus production after Human Immunodeficiency Virus infection of T lymphocytes in the presence of azidothymidine.
[28] Poison By Prescription; The AZT story (1990), by John Lauritsen.
[29] AIDS and the AZT Scandal: SPIN's 1989 Feature, Sins of Omission.
Pfizer's own trial shows it's vaccine increases sickness, and death.
Pfizer's own clinical trial (43,548 participants) has shown that their "vaccine" increases sickness, and death, relative to those in the placebo group. This shows that the Pfizer "vaccine" should have never received emergency use authorization, and that the Pfizer people are a bunch of criminals. This video discuses the trial. 50 MB. Here is an email sized version. 25 MB. There is a PDF that accompanies the video here. The "Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months" article & appendix are here and here. Pfizer's post-marketing pharmacovigilance report mentioned on page 30 of accompanying pdf can be found here. This paper [48] from "Toxicology Reports" looks at the short-comings of the Pfizer trials, and asks the question "Why are we vaccinating children against Covid-19?"
Fuct-checkers have claimed that the animal tests were not skipped as said in the video. However, the animal tests were skipped, and Pfizer proceeded to the human trials without them. The human clinical trials began April 29, 2020, and the animal test results were published on Sept 8, 2020. Animal tests are done to ensure the safety of those in the human trials. That is why animal tests are always conducted before the human clinical trials. That the animal tests were done later made them almost irrelevant.
Some very important developments.
Fully-vaccinated infected at twice the rate of the un-vaccinated.
Statistics from the UKHSA (United Kingdom Health Security Agency) tell us that the fully-vaccinated are being infected at (more than) twice the rate of the un-vaccinated. This has been reported to be the case in every NHS "Covid-19 vaccine surveillance report" since early September 2021. The relevant statistics are reported weekly, and cover the proceeding month.
For example, in the Covid-19 vaccine surveillance report – week 44 (Oct. 4 to 31, 2021) data supporting this conclusion can be found in Table 5 on page 20. The table is titled; "Unadjusted rates of Covid-19 infection, hospitalisation and death in vaccinated and unvaccinated populations." The first few columns of Table 5 are;
Age Cases of Covid-19 Cases of Covid-19
per 100,000 of the per 100,000 of the
fully-vaccinated un-vaccinated
<18 346 2,891
18-29 614 710
30-39 1,221 893
40-49 2,125 933
50-59 1,409 656
60-69 999 460
70-79 741 373
≥80 480 375
Let me spell out what these numbers are saying;
If you are fully-vaccinated, and between 40 and 80 years old, you are about twice as likely to catch covid-19 as the un-vaccinated.
If you are fully-vaccinated, and between 30 and 40 years old, you are 1.4 times as likely to catch covid-19 as the un-vaccinated.
If you are fully-vaccinated, and older than 80, you are about 1.3 times as likely to catch covid-19 as the un-vaccinated.
But, how can a fully-vaccinated individual be more than twice as likely to catch covid-19 as an un-vaccinated individual? Even if the vaccine didn't work, at all, you would only expect about the same number of covid-19 cases in both the vaccinated, and un-vaccinated. But, we see twice the number of covid-19 cases in the fully-vaccinated.
So, what is happening?
Well, this is vaccine dependent enhancement (also called antibody dependent enhancement or ADE). Vaccine dependent enhancement has been observed in all the previously produced vaccines against coronaviruses. This is why no coronavirus vaccine has ever been offered to the public, previous to Covid-19. This is the vaccine dependent enhancement that so many scientific papers, and experts in the field, warned vaccine makers to watch for. But while claiming to be the science, the evil nutters refused to listen to the science. The nutters produced vaccines, and never bothered to check for vaccine dependent enhancement.
But, how can the exact same covid-19 virus be much more infectious after vaccination, than it was before vaccination?
In vaccine dependent enhancement, some antibodies, produced in response to the vaccine, open up a new way for the Covid-19 virus to get into (i.e., infect) your cells. If this new route into your cells is more easily negotiated than the original route, then the virus will be more infectious. If the new route is twice as easy to negotiate, then the virus will be twice as infectious as it was. Even if the vaccine is 100% effective in stopping entry by the original route, you can still be twice as likely to catch the disease due to this alternate, antibody dependent, route into your cells.
It is apparent that the people running the UKHSA website have noticed these figures, and are taking steps to hide them. From week 47 on, the Covid-19 vaccine surveillance reports actually present the offending numbers in grey, so that they will be less noticeable. They also appear to be manipulating/falsifying the data in the age group ≥80. There does not appear to be any rational explanation for the changes occurring in this age group. If you have an explanation, please let me know.
You can find the weekly Covid-19 vaccine surveillance reports here.
The relevent pages from the week 44 report can be found here.
It appears that the vaccine manufacturers, and their owners, only care about taking your money. They did not even care enough about your health to check for vaccine dependent enhancement, which they had to know was likely to occur.
What is antibody-dependent enhancement?
When your body is alerted to a virus, it produces antibodies against it. These are large Y-shaped molecules that have variable regions in the tips of the arms of the Y. Different amino acid sequences in the tips, form different antibodies, that are able to bind to different regions/parts of the virus. Billions of copies of each of these distinct antibodies are created, and this mass of antibodies circulates in the body, with each looking for the piece of virus that it matches, and can bind to. On finding the requisite piece, it binds to it, tagging the virus, so that specialist cells can locate, and destroy, it.
One such specialist cell is the macrophage. These have a (surface) receptor, called an Fcγ-receptor, that attaches to antibodies that are attached to a virus. The tips of the antibody attach to the virus while the lower section of the antibody attaches to the Fcγ-receptor of the macrophage. Once attached, the macrophage engulfs the virus + antibody + Fcγ-receptor, i.e., the macrophage wraps the virus + antibody + Fcγ-receptor in a membrane, and brings it inside the cell, as a bubble, called an endosome. Hydrogen ions, and proteases (enzymes that cut up proteins) are pumped into the endosome, turning it into a acid bath where the virus is destroyed (i.e., broken down into harmless bits and pieces). Generally, this works, the virus is digested, and its bits, and pieces, recycled.
However, this cleanup process does not always work well for the covid virus. The reason being that the covid virus is able to escape endosomes, and infect the surrounding cell. When hydrogen ions, and proteases, are pumped into the endosome to digest the virus, the acidic environment enables a protease, called cathepsin-L, to prime the spikes of the virus. Now, if one of these primed spikes contacts the endosome membrane, it will harpoon the membrane, and merge both viral, and endosome membranes. This dumps the viral RNA into the cell cytoplasm, infecting the cell. For some reason the covid virus cannot productively reproduce in most macrophage cell types, however, in vulnerable individuals, its presence results in a cytokine storm. It is this cytokine storm that kills many people.
Whether or not this cleanup process works for the covid virus depends on how strongly the various antibodies cling to the virus. When enough antibodies cling to the spike proteins, and cling for a long enough time, their physical presence prevents cathepsin-L from priming the spikes, and even if a few spikes are primed, the antibodies attached to other spikes physically prevent them from approaching the endosome membrane, and infecting the surrounding cell. Antibodies that cling to the virus long enough to prevent infection of the surrounding cell, are called neutralizing antibodies.
However, there are also non-neutralizing antibodies. Some antibodies do not attach to the virus strongly, and can dissociate from it before it is disabled. If enough antibodies dissociate from the virus, cathepsin-L is able to prime some of the spikes, and some of these may then push up against the endosome membrane, merge with it, and infect the surrounding cell. Such antibodies are called non-neutralizing antibodies. Non-neutralizing antibodies are dangerous as they open up a new way for the covid-virus to infect your cells. This opening up of a new route of infection, is called antibody-dependent enhancement (of infection) or ADE.
It is worth noting that hydroxychloroquine prevents the acidification of endosomes, which prevents the action of cathepsin-L, and thus may prevent ADE in covid. However, in [45] it is shown that antibody dependent enhancement of SARS-CoV-1 infection occurs in the presence of cathepsin-L inhibitors. This implies another protease (present in the endosomes of macrophages, etc) can prime SARS-CoV-1. Since it is likely that this protease will also prime SARS-CoV-2, it is likely that hydroxychloroquine, by itself, will not prevent ADE in covid.
Here is a diagram from [44]. It illustrates two types of ADE. One being via the Fcγ-receptor (called FcR in the diagram), the other being via the C3b-receptor. Macrophages, monocytes, and dendritic cells have Fcγ-receptors. The Y-shaped molecules represent antibodies. They are labeled IgG. Ig stands for immunoglobulin (another name for an antibody). IgG is the most common variety of antibody. One of the pathways leading to covid-virus infection is illustrated on the left. The pathway via endosomes is strangely missing. The ACE2 (angiotensin-converting enzyme 2) receptor is represented by an orange oval. Outside the cell is brown, inside is green.
Regarding the missing infection pathway, via endosomes, one has. The covid-virus infects a cell by first attaching to a particular cell receptor, called an ACE2-receptor. In the presence of certain proteases, the viral membrane merges directly with the cell membrane, and the viral RNA is dumped into the cell cytoplasm, infecting the cell. In the absence of these proteases, the virus + receptor is bought into the cell in an endosome. Hydrogen ions, and proteases, are pumped into the endosome to digest the virus. Here the protease cathepsin-L primes the spikes of the virus, and when one of these contacts the endosome membrane, it harpoons the membrane, and merges both viral, and endosome membranes. Again, this dumps the viral RNA into the cell cytoplasm, infecting the cell. This process is quite similar to that described above.
The process of a cell enveloping a virus attached to a receptor with a membrane, and bringing it inside the cell as a endosome, is known as receptor-mediated endocytosis. Below, I picture two examples of receptor-mediated endocytosis (Fcγ and ACE2).
Inside an endosome is brown. Inside the cell, but outside the endosomes, is green. The middle diagram illustrates the situation with neutralizing antibodies. The antibodies physically prevent the spikes from being primed, and keep the spikes away from the endosome wall. The diagram on the right illustrates the situation with non-neutralizing antibodies. Here a few antibodies have separated from the virus, and allowed some spikes to be primed. These spikes have approached the endosome wall, and will shortly merge the viral, and endosome membranes, infecting the cell. The left diagram illustrates the situation after ACE2-mediated endocytosis. Here, there nothing to prevent cathepsin-L from priming the spikes, and nothing to prevent the spikes from merging the viral, and endosome membranes, infecting the cell.
For the covid-virus, it should be obvious that even neutralizing antibodies will lead to ADE, if there are not enough of them. Thus the waning of antibody levels will leave you liable to covid infection, and possibly open to more serious disease. For Fcγ-mediated endocytosis to work (against viruses that can escape endosomes) there needs to be a generous coating of the spike proteins by antibodies. Generally, whether ADE occurs, or not, depends on the balance between the concentrations of neutralizing antibodies, and non-neutralizing antibodies.
Fully-vaccinated now infected at triple the rate of the un-vaccinated.
The data keeps on getting worse and worse. If you are between 18 and 80 years of age, and fully-vaccinated, then you are more than three times as likely to catch Covid-19 as someone who is not vaccinated.
Age Cases of Covid-19 Cases of Covid-19
per 100,000 of the per 100,000 of the
fully-vaccinated un-vaccinated
<18 894 1,062
18-29 2,178 722
30-39 2,501 723
40-49 2,241 642
50-59 1,703 496
60-69 1,340 370
70-79 972 356
≥80 1,055 549
The data is from Table 13 of week 10 of the British Covid-19 vaccine surveillance report. It covers the period 7 Feb to 6 Mar, 2022.
Fully-vaccinated now infected at four times the rate of the un-vaccinated.
The data keeps on getting worse and worse. If you are between 18, and 80 years old, and fully-vaccinated, then you are about four times as likely to catch Covid-19 as someone who is not vaccinated.
Age Cases of Covid-19 Cases of Covid-19
per 100,000 of the per 100,000 of the
fully-vaccinated un-vaccinated
<18 1,454 1,711
18-29 3,118 941
30-39 4,324 1,085
40-49 3,957 955
50-59 3,303 779
60-69 2,814 572
70-79 2,161 532
≥80 2,023 775
The data is from Table 14 of week 13 of the British Covid-19 vaccine surveillance report. It covers the period 28 Feb to 27 Mar. Wow.
An amazingly blatant fraud.
Week 3, 2022, of the surveillance report introduces an amazingly blatant fraud. Here, manipulation of the data presented in Table 11(b) reduces the number of covid-19 deaths reported among vaccinated individuals by a minimum of 48%. The fraud is effected by splitting the reported deaths into two groups, and simply ignoring the deaths in one group.
In the age group 18-29 the fraud reduces 9 deaths to 1 death (a 89% reduction in deaths)
In the age group 30-39 the fraud reduces 40 deaths to 6 deaths (a 85% reduction in deaths)
In the age group 40-49 the fraud reduces 59 deaths to 11 deaths (a 81% reduction in deaths)
In the age group 50-59 the fraud reduces 186 deaths to 44 deaths (a 76% reduction in deaths)
In the age group 60-69 the fraud reduces 418 deaths to 140 deaths (a 67% reduction in deaths)
In the age group 70-79 the fraud reduces 751 deaths to 325 deaths (a 57% reduction in deaths)
In the age group ≥80 the fraud reduces 1882 deaths to 982 deaths (a 48% reduction in deaths)
Previous to Week 3 the last column of Table 11(b) was the 7th column and it was labeled "Second dose ≥14 days before specimen date". After Week 3, what would have previously been the 7th column was divided into two columns, columns 7 and 8. These were labeled "Second dose ≥14 days before specimen date" and "Third dose ≥14 days before specimen date". Using the Week 3 data we can easily reconstruct the last column of Table 11(b) as it would have been before the fraud. We obtain;
Age
18-29 9 = (8 + 1)
30-39 40 = (34 + 6)
40-49 59 = (48 + 11)
50-59 186 = (142 + 44)
60-69 418 = (278 + 140)
70-79 751 = (426 + 325)
≥80 1882 = (900 + 982)
From this data the number of deaths, per 100,000, among the vaccinated was calculated for each age group, and recorded as the second to last column of Table 12. This column is next to the last column of Table 12, which records the number of deaths, per 100,000, among the un-vaccinated, for the purpose of comparison.
With the fraud in operation the number of deaths among "suitably triple jabbed" individuals, now reads:
Age
18-29 1
30-39 6
40-49 11
50-59 44
60-69 140
70-79 325
≥80 982
The deaths of vaccinated individuals who are "suitably double jabbed" but not "suitably triple jabbed", i.e., the deaths recorded in the new column 7, are simply ignored, even though they form the majority of the deaths. As before, the last (the 8th) column of Table 11(b) is fed into the second to last column of Table 12, where the (now much reduced) numbers of deaths, per 100,000, among the vaccinated, are compared with the number of deaths, per 100,000, among the un-vaccinated.
This fraud is quite outrageous. In fact, this fraud is so egregious that those responsible for it should be fired and never allowed to work in the public sector again.
Weeks 2, and 3, 2022, of the surveillance report can be found here and here.
Tables 11(b) and 12 from Week 2 can be found here.
Tables 11(b) and 12 from Week 3 can be found here.
Like I said, an amazingly blatant fraud.
The "vaccines" now increase sickness and death for everyone.
In particular, this is true for those over 80 years old.
The following data is, again, from the United Kingdom Health Security Agency (UKHSA).
Lets look at the (covid-19) death data for those ≥80 years of age, from week 3, 2022 (the fraud) onwards:
. Deaths per 100,000
A B C
week03 39 309 270
week04 57 322 265
week05 78 326 248
week06 103 324 221
week07 114 280 165
week08 120 243 124
week09 120 190 70
week10 110 152 42
week11 101 141 40
week12 90 134 44
week13 84 122 37
Deaths per 100,000 means deaths per 100,000 per 28 day period ending with the stated week.
Column A is deaths among those "suitably triple jabbed" per 100,000 per 28 day period.
Column B is the deaths among the un-vaccinated per 100,000 per 28 day period.
Column C is Column B minus Column A.
I must emphasize that the above data only records deaths due to covid-19. It does not record any deaths due to adverse reactions to the vaccines.
So Column C is the number of deaths that are prevented for every 100,000 people who are vaccinated. If this number is negative then it records the number of deaths caused by the vaccines (per 100,000 vaccinated).
Graphing Column C we can see that as it approaches zero it levels off (from week 10 onwards). This is due to those presenting the data finding some fraud to keep the data positive, or simply making up the data. Anything, to keep the data from proving that the vaccines are killing more than they are saving.
Anyway, after manipulating the data for the weeks 10, 11, 12, and 13, the evil people decided that the data was henceforth always going to show that the vaccines are killing more than they are saving, so they stopped publishing the data. To provide an excuse for this they had to arrange for the UK Government to stop paying for covid-19 testing, so that is what they did. In the week 14 surveillance report they state:
From 1 April 2022, the UK Government ended provision of free universal covid-19 testing for the general public in England, as set out in the plan for living with Covid-19. Such changes in testing policies affect the ability to robustly monitor covid-19 cases by vaccination status, therefore, from the week 14 report onwards this section of the report will no longer be published.
Actually, it is obvious they are lying to you. For hospitalization, and death, their claim is clearly false, as the patient's names can easily be matched against vaccination records to provide the data, just as before. However, a partially true excuse is better (to deceive you) than no excuse at all.
Remember, the evil people's job is to lie to you. To lie to you in such a way that you believe the lies are the truth.
The relevant pages from the covid-19 surveillance reports can be found here.
Previously, this very data was used to justify the vaccine mandates. The argument was that the vaccines are saving more than they are killing. This is no longer true. What is now true, is that;
The vaccines are killing more than they are saving.
As time goes on the vaccines are likely to kill far, far more, than they save. Exactly how many more will not be known as they have chosen to stop reporting the data.
The 2020 lockdowns cost some 160,000 American lives.
The economic cost of the lockdowns has been huge. The number of lives lost to the lockdowns has been enormous. Here we estimate the number of United States lives lost to the lockdowns during the year 2020. Below are the weekly non-covid deaths for 2019 and 2020 (from late March) as recorded by the CDC (Centers for Disease Control and Prevention).
----------------------
2019 NON-COVID 2020
DEATHS
----------------------
3/30 56675 59990 3/28
4/6 56598 62660 4/4
4/13 55491 63585 4/11
4/20 54468 60512 4/18
4/27 53656 59254 4/25
5/4 53987 56864 5/2
5/11 53478 56355 5/9
5/18 53578 55986 5/16
5/25 53695 55034 5/23
6/1 52727 54090 5/30
6/8 53141 54392 6/6
6/15 52646 54299 6/13
6/22 52290 54650 6/20
6/29 52209 55173 6/27
7/6 52344 55774 7/4
7/13 51931 56708 7/11
7/20 51649 56646 7/18
7/27 51662 56725 7/25
8/3 51410 56624 8/1
8/10 51747 56535 8/8
8/17 51023 57083 8/15
8/24 51022 56822 8/22
8/31 51162 55957 8/29
9/7 51836 55812 9/5
9/14 51633 55587 9/12
9/21 51757 55975 9/19
9/28 52757 56821 9/26
10/5 52564 56111 10/3
10/12 53090 57501 10/10
10/19 54338 56053 10/17
10/26 54049 56862 10/24
11/2 54087 57172 10/31
11/9 55699 59744 11/7
11/16 55886 59244 11/14
11/23 56199 59612 11/21
11/30 55465 59171 11/28
12/7 57303 60573 12/5
12/14 57658 62941 12/12
12/21 57424 62512 12/19
12/28 58458 63003 12/26
-----------------------
2,148,792 2,306,412
-----------------------
Each date is a Saturday.
The second column is the number of non-covid deaths for the week (in 2019) ending on the date in the first column.
The third column is the number of non-covid deaths for the week (in 2020) ending on the date in the fourth column.
The non-covid death numbers for 2019 are just the usual morbidity numbers (covid played no part here).
The 2019 non-covid death numbers are as provided by the CDC here.
The 2020 non-covid death numbers are as provided by the CDC here.
The last row is the totals of the columns 2 and 3.
Some deaths in Dec 2020 will be vaccine related.
From this one can conclude that the 2020 lockdowns caused around 2,306,412 - 2,148,792 = 157,620 extra deaths in the U.S.
Consider also, that many of the covid deaths were in reality non-covid deaths.
The Covid-19 Scam & Vaccines.
This used to be (and will be again) the top of the article.
A new section on the Covid-19 vaccines can be found below.
The latest VAERS (Vaccine Adverse Event Reporting System) data, repackaged to make it readable, can be found here. (WinZip opens .gz files)
This file lists 77,827 Covid-19 sequences and their mutations.
The Covid-19 HOAX can be seen in the way Covid-19 spread.
It spread to the whole world but jumped over the major Chinese cities.
You know Shanghai, Beijing, Guangzhou, Hong Kong, etc. On March 16, about 2 months after the Wuhan lockdown,
Beijing Municipality had only 442 confirmed cases of Covid-19 (population 20 million),
Shanghai Municipality had only 353 confirmed cases of Covid-19 (population 23 million),
Guangdong Province had only 1,357 confirmed cases of Covid-19 (population 104 million),
Hong Kong Region had only 141 confirmed cases of Covid-19 (population 7 million).
Get that... it didn't appreciably spread (before or after the Wuhan lockdown) to any of the major Chinese cities.
But there was a tremendous spread of the disease (before the Wuhan lockdown) to Iran and Italy.
How's that?
The Washington Post reported that 5 million people left Wuhan between January 10 (the start of the Chinese New Year travel rush) and the lockdown.[1]
Get that... at the height of the uncontrolled epidemic, five million leave Wuhan for elsewhere in China, but do not appreciably spread the disease. They spread it to many other cities, but very few catch the disease.
But a very small number from Wuhan, somehow, massively spread the disease to Iran and Italy.
How's that?
Already we have very strong evidence that Covid-19 was deliberately spread. The disease was observed to spread rapidly throughout Wuhan indicating it was very contagious, yet millions left Wuhan for the new-year celebrations in their ancestral towns, and spread the virus throughout the whole of China, but very few caught the disease, indicating the disease is hardly contagious at all. Then it suddenly becomes very contagious in Iran, and Italy.
And what about Africa?
As of April 16, there were only 16,500 confirmed cases of Covid-19 in all of Africa.
Get that... only 16,500 cases in all of Africa. Africa, which has seen massive Chinese investment accompanied by over a million Chinese workers.
How's that?
And what about the spread of the Delta variant. What good are the lockdowns, travel restrictions, and isolation. The Delta variant charges right past them all. Do they think we are stupid? This is further very strong evidence that Covid-19 is being deliberately spread. (Actually, since the PCR tests do not differentiate the Delta variant, it is possible that a great many cases are simply being labeled the Delta variant, whether they are or not. In this way the "breakthrough cases" can be blamed on a scary new variant, rather than admit that the vaccines simply do not work as effectively as claimed against the original varieties.)
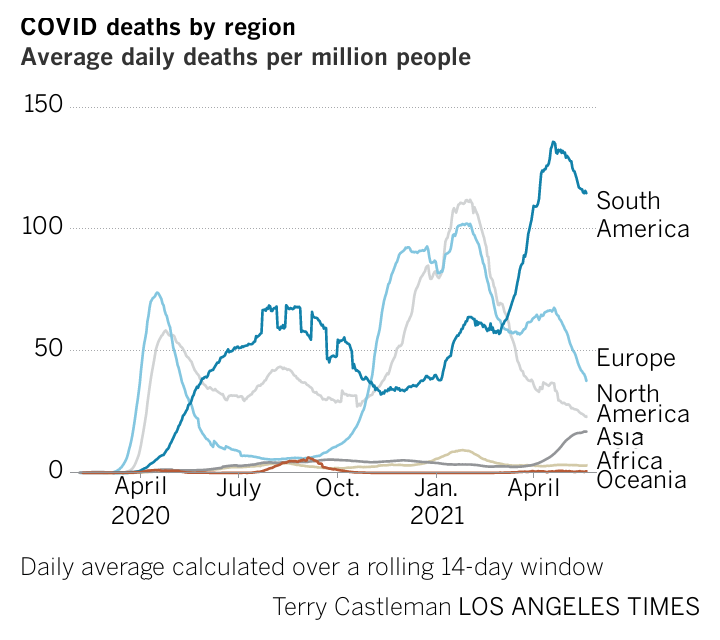
The above graphic from the Los Angeles Times shows the daily Covid-19 deaths (14 day average) per 100,000 people, for each continent. Note that Covid-19 was never much of a problem in Africa and Asia, even though the RNA data showed that it had been found right across Africa, and Asia, by March/April 2020. This is incredibly strong evidence that Covid-19 has been deliberately spread (mainly to the rich countries).
Notes: Guangzhou is the capital of Guangdong Province.
See below for the March 16 record of the confirmed cases in each of the Chinese Provinces/Municipalities.
The Wuhan lockdown occurred on January 23. All flights from Wuhan stopped on January 23. Flights resumed on April 8 when the Wuhan lockdown was lifted. With the opening on April 8 you were free to visit China and see everything for yourself. Two weeks of isolation was required upon entry.
So the early evidence showed that Covid-19 is not particularly contagious.
However, the media pushed the story that Covid-19 was both very contagious, and very dangerous.
Since the beginning, there has been a coordinated effort to exaggerate the dangers of Covid-19.
This has been admitted by officials in Italy, and England.
"But Prof Ricciardi (Scientific Adviser to Italy's Minister of Health) added that Italy's death rate may appear higher because of how doctors record fatalities. "The way in which we code deaths in our country is very generous in the sense that all the people who die in hospitals with the coronavirus are deemed to be dying of the coronavirus." From here or here.
Prof Ricciardi also said: "On re-evaluation by the National Institute of Health, only 12 per cent of death certificates have shown a direct causality from coronavirus (Covid-19), while 88% patients who have died have at least one pre-morbidity – many had two or three."
The UK's Chief Scientific Officer, Sir Patrick Vallance, stated: "It is worth remembering again that the ONS (Office for National Statistics) rates are people who've got Covid on their death certificates. It doesn't mean they were necessarily infected because many of them haven't been tested. So we just need to understand the difference."
Policies designed to spread Covid-19 to Care Homes.
On March 25th, 2020, New York Governor Cuomo issued an executive order forcing the transfer over 4,500 Covid-19 patients from hospitals to their nursing homes (also called old-age or care homes). In the following weeks the nursing homes saw 6,000 deaths from Covid-19. The decision drew fire as soon as it was announced from medical experts, nursing home operators, and the families of residents. However, it was forced through anyway. Similar orders were given in a number of other states. See here.
When the Justice Department requested data (26 August 2020) from governors Andrew Cuomo of New York, Phil Murphy of New Jersey, Tom Wolf of Pennsylvania and Gretchen Whitmer of Michigan, on their orders requiring nursing homes to admit Covid-19 patients, Cuomo and Whitmer dismissing it as a political charade saying that "At least 14 states - including Kentucky, Utah and Arizona - have issued similar nursing guidance all based on federal guidelines...." See here.
Playing Russian roulette: Nursing homes told to take the infected. California, New Jersey and New York have made nursing homes accept Covid-19 patients from hospitals. Residents and workers fear the policy is risking lives. That is the title of this New York Times article.
You could make a strong argument that the USA's deadly coronavirus problem is largely a nursing home problem, dangerous everywhere but far more prevalent in a half-dozen or so of the country's more heavily and densely populated states. What's more, many of these states enacted coronavirus response policies that likely put nursing home residents at higher risk for infection. See here.
In mid-June 2020, Republican Whip Steve Scalise announced that he had sent letters to the governors of California, Michigan, New Jersey, New York, and Pennsylvania, demanding they explain why they ignored protocols and forced Covid-19 patients into nursing homes. Scalise noted, "While nursing home residents make up 0.6% of the U.S. population, they account for 42% of nationwide Covid-19 deaths". That is, 70% of all U.S. Covid-19 deaths had (up till that time) been in care homes.
As of Nov. 24, 2020, it was claimed that 264,000 people in the United States had died from Covid-19. Of these deaths 100,033 occurred in nursing homes (long-term care facilities). That is, 38% of all U.S. Covid-19 deaths had (up till that time) been in care homes. The rest of the U.S. was beginning to catch up.
The Amnesty International Reports: Covid-19 and Care Homes.
Policies designed to kill hundreds of thousands of the elderly.
The report on the United Kingdom. Some passages from the report:
Covid-19 has had a devastating impact on older persons living in care homes in England. 28,186 "excess deaths" were recorded in care homes in England between 2 March and 12 June, with over 18,500 care home residents confirmed to have died with Covid-19 during this period. UK government decisions and failures resulted in violations of the human rights of people living in care homes, notably the right to life, to health and to non-discrimination. From discharging 25,000 patients, including those infected, into care homes; to denying care homes residents admission to hospital and imposing "do not attempt resuscitation" orders on them without due process, to failing to provide PPE (personal protective equipment) and testing to care homes. Older persons living in care homes were abandoned to die.
I guess; "people in care homes had their right to life violated", sounds better than; "people in care homes were murdered," but it is less accurate.
The following was so deceptively worded that I have added explanation (in brackets).
The Department of Health and Social Care.... adopted a policy,... that led to 25,000 patients, including those (known to be) infected (with Covid-19, and also those who were) possibly infected with Covid-19 (as they) had not been tested, being discharged from hospital into care homes between 17 March and 15 April—exponentially increasing the risk of transmission to the very population most at risk of severe illness and death from the disease. (This, while being denied) access to testing, (being denied) personal protective equipment, (while having) insufficient staff, and limited (and confusing) guidance. (As expected) care homes were overwhelmed.
Care home managers have reported to Amnesty International, as well as to the media, that they were pressured in different ways to accept patients discharged from hospital who had not been tested or who were Covid-19 positive.
I was contacted by concerned residents, saying surely we wouldn't put Covid-19 patients in care homes where the most vulnerable are? But commissioning [the council commissioning department] said, yes we are....
They endangered older people by discharging infected patients into care homes, without even providing tests and personal protective equipment.
Baroness Ros Altmann said: "care homes were left behind in the scramble for PPE, for emergency admission, ventilation and for testing... It's almost as if the system is stacked against them."
Care England has also reported incidents of supplies ordered by care homes being requisitioned for the NHS (National Health Service).
Our local hospital always had over 500 empty beds and so staff were not under pressure and they had lots of PPE. (However) we had 45 percent of the staff self-isolating and were scrambling to get PPE and even food.
By barring both oversight and family visits, the government increased the risk that care home residents would be exposed to abuses that would not be identified, reported and investigated.
It was the same in Italy. Some passages from the Italian report:
We can distinguish the transfers of Covid-19 positive patients discharged from hospitals to care homes:
1) patients from hospitals who were no longer acute but still Covid-19 positive.
2) patients from hospitals who were assumed non-Covid-19 positive, but were not tested,...
Infected patients discharged from hospitals also arrived in the same facilities. Some had not been swabbed at all (i.e., were not tested), but others had been determined positive for Covid-19 in hospital and had been sent to care homes without verifying the homes ability to assist these people safely.
The transfers of patients from hospitals to nursing homes took place continuously, without warnings, instructions or discussions.
The sending of positive Covid-19 patients discharged from hospitals to care homes.... where there were inadequate supplies of nasal swabs, and PPE, endangered the lives of residents and staff.
The managers of a care home reported that it has not been able to obtain any PPE from local health authorities for several weeks and that the small quantity that they eventually managed to buy, independently, was requisitioned by the customs authorities and redirected to hospitals.
Hundreds of patients had died, but in many homes, in Lombardy and elsewhere, swabs were rare, mostly not available at all. In many facilities, the first swabs — both for residents and operators — arrived only in the month of April, when the peak of infections and deaths had passed and thousands of residents had died. In other homes they arrived weeks later than this.
In Lombardy the situation was completely lost: we were abandoned like ships at sea without fuel, a total abandonment, they didn't even answer the phone.
Nobody is going in [to care homes], so there are no witnesses to whatever is going on.
Amnesty International report: United Kingdom (English)
Amnesty International report: Italy (Italian)
Amnesty International report: Spain (Spanish)
Amnesty International report: Belgium (French)
It is interesting that the United States branch of Amnesty International did not see fit to publish anything on the situation there.
The Spanish Amnesty International report is to a large extent damage control. This report has been carefully written to avoid all mention of hospitals sending Covid-19 positive patients to care homes. However, it has been widely reported that Covid-19 spread to care homes more rapidly in Spain than in any other country. And, if the hospitals did not spread the disease to the Spanish care homes, then how did it spread to them so quickly?
That Spanish hospitals discharged positive Covid-19 patients into care homes can be seen from here, section 3.2.2, where it says: "The guide for the prevention and control of Covid-19 in nursing homes and other residential centres indicates that if a resident has been hospitalized they may be returned to their established care home, or newly admitted to a care home, even if their PCR is still positive." The guide goes on to say that the patient should be isolated and monitored in the home for 14 days (but in many cases this was made difficult, if not impossible).
The complete Spanish Amnesty International report includes 5 short videos and 3 PDFs. You can download them here.
The New York Governor, Cuomo, refuses to say who conceived of the order (to send Covid-19 positive patients to care homes). But as you can see, the person/people who gave the order, gave it worldwide. Who has this sort of power? Perhaps Cuomo should be charged with murder to loosen his tongue.
Recently, the media has been trying to impeach Cuomo over the massive under-counting of nursing home deaths, and numerous accusations of sexual harassment. These are designed to throw the real issue into the background. However, Cuomo will eventually have to be thrown under the bus to protect those who actually designed (and understood the implications of) the policy which Cuomo carried out.
Unsurprisingly, those countries known to have adopted this policy (of sending Covid-19 positive patients to care homes) have among the highest number of deaths (per unit of population). It would be interesting to know if the other countries with very high death percentages, namely, Bulgaria, Portugal, Slovenia, and Czechia, also subscribed to this evil policy.
You may ask why the media has refused to tell you about the Amnesty International reports. You may ask why the media is protecting these killers.
Proof that homes for the aged were targeted.
The province of Ontario, Canada, has two types of homes for the aged:
Long-term Care Homes, which are administered by the Ministry of Long-term Care, and
Retirement Homes, which are administered by the Retirement Homes Regulatory Authority.
Retirement Homes are not required to provide the same level of care as Long-term Care Homes, however, many Retirement Homes are, in essence, Long-term Care Homes. Long-term Care Homes are largely government funded, whereas Retirement Homes are not. Long-term Care Homes are 54% privately run, and 46% government run. Retirement Homes are 100% privately run.
There are about 627 (licensed) Long-term Care Homes in Ontario.
There are about 770 (licensed) Retirement Homes in Ontario.
There were Covid-19 outbreaks in 272 (43.4%) of the 627 Long-term Care Homes, till Apr 7.[9]
There were Covid-19 outbreaks in 38 (4.9%) of the 770 Retirement Homes, till Apr 7.[10]
So, the Long-term Care Home outbreaks are 43.4/4.9 = 8.86 times more prevalent than Retirement Home outbreaks.
Because an outbreak is, by definition, the introduction of Covid-19 into an old-age home from the outside, the most important factor influencing an outbreak is the number of people that the residents meet from outside the home. Long-term Care Home residents would generally meet more staff (from outside the home), whereas Retirement Home residents would meet more outsiders while they were away from the home, as they are free to wander wherever they choose. Which of the two groups would meet more outsiders? One suspects that Retirement Home residents would come into contact with more outsiders than Long-term Care Home residents.
Given this, one would expect that Retirement Homes would have had a few more outbreaks than Long-term Care Homes. However, initially, Long-term Care Homes had nearly nine times the number of outbreaks. This is proof that Covid-19 was deliberately spread to Ontario's Long-term Care Homes, but not to the Retirement Homes. It appears that the list of targeted places included Long-term Care Homes, but not Retirement Homes. An administrative oversight by the spreaders?
[9] In the article: Risk Factors Associated With Mortality Among Residents With Coronavirus Disease 2019 (Covid-19) in Long-term Care Facilities in Ontario, Canada, we read "In this analysis, we documented the rapid spread of Covid-19 through Ontario's long-term care system, with a marked increase in risk of death among older residents with frailty during a brief period from late March to early April 2020 (April 7)," and "A total of 627 long-term care facilities were included in the provincial data set; of these, 272 (43.4%) were identified as having either confirmed or suspected Covid-19 infection in residents or staff."
[10] Data for the number of Retirement Homes with Covid-19 outbreaks has been collected by the Retirement Homes Regulatory Authority. It is displayed here. Click on "Timeline of Active & Resolved Outbreaks". Strictly, it should be "There were Covid-19 outbreaks in at most 38 of the 770 Retirement Homes." The data from March to July can be found here.
More evidence that homes for the aged were targeted.
In Ontario, as of June 1, there were:
1,652 (89.2%) deaths in Long-term Care Homes.[11]
199 (10.8%) deaths in Retirement Homes.[12]
So, there were about nine times as many deaths in Long-term Care Homes.
About nine times as many outbreaks has led to about nine times as many deaths.
This indicates that the severity of the outbreaks in both classes of home were similar, which is not what we have been told. Although, this is roughly what one should have expected as Long-term Care Homes, and Retirement Homes, are quite similar.[15]
Some amazing statistics.
As of 1 June, 92% of all Covid-19 deaths in Nova Scotia, occurred in Long-term Care Homes.[13]
As of 1 June, 88% of all Covid-19 deaths in Quebec, occurred in Long-term Care Homes.[13]
As of 1 June, 1,851/2,293 = 81% of all Covid-19 deaths in Ontario occurred in Long-term Care Homes.[13]
These percentages tell us that there were very few cases outside the Long-term Care Homes from which Covid-19 could naturally spread into the Long-term Care Homes. This is more evidence that Covid-19 was deliberately spread to Ontario's Long-term Care Homes. It also shows that in Ontario the disease was not actively spread to the general community. Someone got lazy?
By June 1, the Long-term Care Homes in the Canadian provinces/territories, Prince Edward Island, Yukon, Northwest Territories, and Nunavut had no cases of Covid-19 at all, and those in Newfoundland and Labrador, New Brunswick, Manitoba, and Saskatchewan had 12 cases between them (including 4 deaths).[14]
So, it seems that the spreaders lacked the manpower to tackle all the Care Homes in the more sparsely populated areas of Canada. In fact, the lack of manpower was felt globally. Remember, that as of April 16, months after the beginning of the pandemic, there were only 16,500 confirmed cases of Covid-19 in all of Africa.
[11] From here. The number of resident deaths in Long-term Care Homes, till June 1, is given to be 1,652. Note that the data before April 25 (i.e., the data concerning the first 654 deaths) has been censored.
[12] Table 2, page 7, of the article, Understanding the impact of Covid-19 on residents of Canada's long-term care homes – ongoing challenges and policy responses, states that the number of deaths in both Long-term Care Homes, and Retirement Homes, as of June 1, is 1,851. From [11] we have that the number of deaths in Retirement Homes is 1,851 - 1,652 = 199.
[13] ibid, figure 2.
[14] ibid, table 1.
[15] "Most Ontarians can be excused for confusing retirement homes with long-term care homes, since they both house older adults with care needs. The similarities are numerous: Both provide care to seniors, are regulated by government with regard to care, and have roughly equivalent numbers of homes and residents across the province. While some retirement homes focus on more independent seniors who require less care, others have stepped up their offerings in assisted living and dementia care – some even in palliative care – to become direct substitutes for long-term care homes." From here. The similarity of Retirement Homes and Long-term Care Homes can also be seen from the information presented in the image which is from this web-page.
{kind=link}
The notation describing mutations.
As most know, one's genetic code is made up of DNA, which is basically long sequences of the four chemicals (called nucleotide bases) adenine, cytosine, guanine, and thymine. Such sequences are represented by strings of the four letters A, C, G, and T, where A stands for adenine, C is for cytosine, G is for guanine, and T is for thymine. For example, the very short string CATG represents the DNA sequence, cytosine joined to adenine, joined to thymine, joined to guanine.
The genetic code of most viruses, including the Covid-19 virus, is slightly different. It is made up of RNA which is basically long sequences of the four chemicals, adenine, cytosine, guanine, and uracil. Uracil is an unmethylated form of thymine. So RNA is quite similar to DNA. Because of this similarity, the notation for DNA is often used for RNA, with the understanding that every T in the notation represents the base uracil. As an example, the string CATG represents the RNA sequence, cytosine joined to adenine, joined to uracil, joined to guanine.
Mutations are changes in the DNA, or RNA, sequence. A mutation which involves a simple change of one of the bases for another, is written as the original base, the position of that base in the sequence, and the new base at that position, all run together. For example, the notation A25958G represents a mutation at position 25958 where an adenine base A has been replaced by a guanine base G. Such a mutation is called a single-nucleotide polymorphism, or SNP, for short.
Another type of mutation, called a deletion, is when a number of bases are lost from a sequence. For example, if a sequence has lost the four base string AGTT starting at position 669, then this is denoted AGTT669- where the dash at the right indicates the bases on the left have been lost.
Another type of mutation, called an insertion, is when a number of extra bases are added to a sequence. For example, if a sequence has gained the three base string ATC starting before the position 1075, then this is denoted -1075ATC where the dash at the left indicates the bases on the right have been added. (I am not sure if insertions are traditionally added before, or after, the position. I chose before, as it simplified the writing of a couple of scripts.)
The various clans of the Covid family.
Early in the epidemic (March) it became clear that the Covid-19 family of viruses could be divided into various clans. One of the earliest reports mentions three major clades (groups with a common ancestor) of the family, the G-clade, the V-clade, and the S-clade.
The G-clade comprised viruses with a single amino-acid change D614G in the spike protein.
The S-clade comprised viruses with a single amino-acid change L84S in the protein ORF8.
The V-clade comprised viruses with a single amino-acid change G251V in the protein ORF3.
D614G represents a change of the amino-acid, at position 614, from aspartic acid to glycine.
L84S represents a change of the amino-acid, at position 84, from leucine to serine.
G251V represents a change of the amino-acid, at position 251, from glycine to valine.
The position is from the beginning of the protein mentioned.
In time some authors described the clans by the underlying RNA changes, thus:
The G-clade consisted of viruses with (at least) the mutation A23403G.
The S-clade consisted of viruses with (at least) the mutation T28144C.
The V-clade consisted of viruses with (at least) the mutation G26144T.
A23403G represents the change of a single base, at position 23403, from adenine to guanine.
T28144C represents the change of a single base, at position 28144, from thymine to cytosine.
G26144T represents the change of a single base, at position 26144, from guanine to thymine.
The position is from the beginning of the standard Covid-19 genome.
A23403G is (almost) always found as part of the group of mutations C241T, C3037T, C14408T, and A23403G.
T28144C is (almost) always found as part of the group of mutations C8782T, and T28144C.
G26144T is (almost) always found as part of the group of mutations C14805T, and G26144T.
To put numbers to the last statements we have:
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] (10120)
C8782T [1835] T28144C [1816] (1807)
C14805T [999] G26144T [945] (848)
where the number of individual occurrences (of the mutations), in square brackets, are compared to the group occurrence (of the mutations), in parentheses. These numbers are derived from the collection of 14,712 viral sequences that I acquired in May.
We will call G-clade viruses, the British-American strain, since their geographical distribution is;
| 3083 | United States |
| 3045 | United Kingdom |
| 452 | Denmark |
| 318 | France |
| 309 | Iceland |
| 295 | Australia,... |
We will call the four mutations C241T, C3037T, C14408T, and A23403G the Group of Four. This means that the British-American strain is (essentially) the viruses that have (at minimum) the Group of Four mutations.
About 80% of all Covid-19 viruses carry the Group of Four mutations. This fact is very difficult to explain.
Another way of saying this is that 80% of all Covid-19 viruses carry the D614G variant. Since there is no good explanation of this fact, the media has, as usual, resorted to lies. They claim that D614G makes the virus 7% more transmissible, and "therefore" the D614G variant has displaced the parental Wuhan virus (to the point where there is almost no Wuhan virus left). Of course, this is complete garbage. Except in the individual where D614G supposedly first appeared, the D614G and Wuhan variety will never compete (until most of the population has been infected). They will infect different bodies, and follow separate paths through the population. They do not compete. Contrary to what we are told, the parental Wuhan virus, and D614G, would both flourish. D614G could not, and would not, displace the parental virus. That you have been told it did, just tells you that you have been lied to.
We will call S-clade viruses, the Asian-American strain, since their geographical distribution is;
| 1235 | United States |
| 134 | China |
| 110 | Australia |
| 81 | Spain |
| 40 | Canada |
| 38 | United Kingdom,... |
and most of the early sequences are Chinese. The S-clade is (essentially) the set of viruses containing the mutations C8782T and T28144C. This has a large subgroup consisting of viruses carrying the five mutations C8782T C17747T A17858G C18060T and T28144C. These five mutations will be called the Group of Five. We will call viruses with these mutations, the American strain, as sequences which carry these five mutations (and no more) are entirely North American. We have:
C8782T [1835] C17747T [1158] A17858G [1177] C18060T [1188] T28144C [1816] (1146).
We will call V-clade viruses, the British strain, since their geographical distribution is;
| 458 | United Kingdom |
| 98 | United States |
| 70 | Australia |
| 69 | Iceland |
| 39 | Netherlands |
| 19 | Greece,... |
The V-clade is (essentially) the set of viruses containing the mutations C14805T and G26144T. This has a large subgroup consisting of viruses carrying the three mutations G11083T C14805T and G26144T. These three mutations will be called the Group of Three. We have:
G11083T [1477] C14805T [999] G26144T [946] (814).
I repeat a line from above as there is much to learn from it:
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] (10120)
This tells you that:
The mutation C241T occurs in 10258 sequences (out of the 14712),
C3037T occurs in 10283 sequences,
C14408T occurs in 10283 sequences,
A23403G occurs in 10328 sequences, and
the group [C241T C3037T C14408T A23403G] occurs in 10120 sequences.
Since the mutations C241T, C3037T, C14408T, and A23403G are widely distributed over the genome they could not have all been added to the genome at the same time. However, since the number of individual occurrences of each mutation are almost the same, all four mutations had to have been added at roughly the same time. This contradiction means that the Group of Four mutations did not occur naturally.
This proves that the British-American viruses are not descendant from the Wuhan variety. Which means that the British-American viruses are a separate occurrence of Covid-19. This means that both occurrences of the virus were deliberate releases, and that both are, almost certainly, engineered. Interestingly, an effort was made to derive the Group of Four from the Wuhan sequence by faking intermediate sequences. You can read about this below.
An impossibly high rate of evolution.
In early April the data from nextstrain.org showed some 4,000 varieties of Covid-19 worldwide.
That so many varieties developed in such a short time, is impossible.
In early May I acquired a collection of 14,712 complete sequences. These sequences contained 8,047 distinct mutations.
Using the evolutionary substitution rate of 24 nucleotide substitutions, per year (i.e., 8 x 10^-4 subs/site/year), and assuming an exponential growth model, the time from the start of the epidemic can be estimated from the equation:
(24)^x = 8,047.
Solving this one finds that it would take x = 2.83 years to accumulate 8,047 mutations, so it seems the epidemic actually started a couple of years before Wuhan. How is that possible?
Was it possible that a strain of Covid-19 had been circulating for some time before Wuhan. If the disease of the strain was mild, then it would pass as the flu, or a cold, and could become widespread without being noticed.
If a strain of Covid-19 did precede Wuhan, then its disease was necessarily mild, in which case there is the intriguing possibility that exposure to it may endow resistance to the more dangerous Wuhan strain. If true, then the less virulent strain of Covid-19 could be a natural vaccine against the Wuhan strain, just like cowpox was a natural vaccine against smallpox. That this might be the case is suggested by the large number of people that appear to be naturally resistant to the virus. These people being resistant due to their previous exposure to the less virulent strain.
There was some indirect evidence that the British-American strain of Covid-19 may have predated Wuhan. However, its disease was unlikely to be mild. Certain papers, see here, here, and here, suggested that the British-American strain was probably more contagious, and more dangerous. This being because the British-American mutation A23403G produces a D614G change in the Spike protein that facilitates viral entry into human cells (via the ACE2 receptors). Since there is no direct evidence that any strain of Covid-19 preceded Wuhan, this possibility has to be discounted.
My latest Covid-19 collection has 77,827 sequences, and is from September, 2020. These sequences contain 23,693 distinct mutations. Let's calculate the time to the beginning of the epidemic from the equation;
(24)^x = 23,693.
Solving this one finds that it would take x = 3.17 years to accumulate 23,693 mutations. This places the beginning of the epidemic at about 3.17 years before September, 2020, which is 3.17 - 0.33 = 2.84 years before May 2020, which agrees with the previous estimate, 2.83. [The 0.33 years is the 4 months between May and September. The estimates here are quite rough, and that they agree so closely, is surprising.]
It should be noted that if a strain is not particularly contagious, then it is much, much easier to control.
In fact, it turns out that the Wuhan strain itself is not particularly contagious. But, if the Wuhan strain is not particularly contagious, then how did it spread so rapidly in Wuhan? Simple, in Wuhan the virus was deliberately spread. It did not appreciably spread to the rest of China simply because it was not particularly contagious. Remember, that at the height of the uncontrolled epidemic (Jan. 10 to Jan. 23) five million people from Wuhan traveled to all parts of China (for the New Year celebrations on Jan. 24) but did not appreciably spread the virus. They spread the virus throughout China, but few caught the disease.
Did Iceland have a strain of Covid-19 before Wuhan?
Between January 31 and April 1 the Icelandic company deCODE Genetics sequenced viral RNA taken from 643 Icelanders who had tested positive for Covid-19. They presented 601 sequences to GISAID. By mid-April, 41 sequences had been removed, leaving 560 sequences, 231 of these being distinct. These 231 sequences contain 326 distinct mutations, of which, 124 are unique to Iceland. [Nextstrain.org originally displayed more than three hundred of these genomes from Iceland, but has since purged all but twelve of them.]
The most commonly occurring of these native Icelandic mutations are: C5142T with 47 cases, A25958G with 29, A1321C 18, T9445C 14, T17531C 10, A4140G 10, T3034C 9, T17859C 9, and G27430A with 8.
On Apr 15, 2020, Kari Stefansson, the CEO of deCODE Genetics, stated that "Currently 291 mutations have been found in the country that have not been identified elsewhere."
That the 291 native mutations established themselves in the viral population in a few weeks is impossible to believe. Mutations/errors may be common but those that establish themselves in the population are not.
291 mutations in Iceland in 2 months is about
= 300 mutations/350,000 people/2 months
= 1,800 mutations/350,000 people/year
= 360,000 mutations/70,000,000 people/year
= 360,000 mutations in Great Britain in one year.
Does that sound likely to you?
This essentially proves that the Covid-19 virus already existed in Iceland prior to Wuhan. Or does it?
The existence of the Group of Four proves that Covid-19 is not of natural origin. After their production the viruses appear to have been kept alive in cell cultures in vials or flasks. Thus the viruses would gather mutations as time passed. This means that the epidemic would date to the production date, not the release date. This explains why the beginning of the epidemic seems to date a couple of years too early. This would also explain the explosion of variant genomes. What one was seeing was the uncovering in weeks, of mutations that had occurred over years, while the viruses were being tampered with, while awaiting eventual release.
The main clades were not due to random mutations but resulted from differences in engineering the viruses. After the engineered viruses were added to cell cultures natural random mutations would have accumulated. Thus when the disease was spread, it was spread as a cocktail of different Covid-19 variants. Each vial would have had its own signature mutations.
French man had Covid-19 before the Chinese.
Amirouche Hammar, a resident of Bobigny, Paris, fell sick with an influenza-like illness. On 27 December 2019, after 4 days of dry cough, fever, fatigue, and breathing difficulties, he drove himself to the emergency ward of the local hospital, where he was administered antibiotics, and recovered sufficiently to be able to leave on December 29. Like all respiratory samples, that of Hammar was frozen, and stored, for possible future analysis. Between 6-9 April 2020 fourteen of these stored samples were tested for Covid-19. The sample of Hammar, taken on 27 December 2019, tested positive for Covid-19. This means that he caught the disease sometime around 12 December 2019, right up there with the very first Chinese cases in Wuhan. The first Wuhan sample was collected on 24 December 2019.
You can read more about it in the paper SARS-CoV-2 was already spreading in France in late December 2019, in the "International Journal of Antimicrobial Agents," which can be downloaded from the internet or here.
The case of Hammar remains a mystery to the scientific community.
The Munich cluster. A complete fabrication.
The paper "Investigation of a Covid-19 outbreak in Germany resulting from a single travel-associated primary case: a case series," from "The Lancet Infectious Diseases," has to be the worst "peer-reviewed" article I have come across. With 41 authors, and at least 1 referee, you would assume that any obvious mistakes would have been weeded out, but this was not the case. It is so obviously wrong that its intention may have been, not to inform, but to mislead.
The article claims to document a case where Covid-19 was spread from China to Europe.
The carrier of Covid-19 was Patient 0, a Chinese business woman who traveled from Shanghai to Munich on Jan 19, 2020, to facilitate workshops, and attend meetings with her German colleagues. She showed no signs of illness during her stay in Germany, although she mentions suffering from jet-lag. She returned to China on Jan 22 where she became ill, and on Jan 26 tested positive for Covid-19. It is claimed that, although asymptomatic at the time, she passed the disease to a number of German associates, who then passed it to others. Those who tested positive for Covid-19 (by qRT-PCR) had their virus sequenced. The sequences were submitted to GenBank. Those infected were called the Munich, or Bavarian, cluster.
One of those infected, Patient 1, is claimed to have caught the disease from Patient 0. Patient 1 then passed the disease to Patient 3. Since Patient 3's only contact was with Patient 1, this certainly appears plausible, however, this turns out to have been impossible.
Consider the mutations (changes from the standard Covid-19 sequence from Wuhan) of Patients 1 and 3.
Patient 1, MT270101.1, SARS-CoV-2/human/DEU/BavPat1-ChVir929/2020, complete genome.
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- (deletion at beginning)
C241T (substitution at position 241)
C3037T (substitution at 3037)
A23403G (substitution at 23403)
CCATGTGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29837- (deletion at end)
Patient 3, MT270103.1, SARS-CoV-2/human/DEU/BavPat3-ChVir1020/2020, complete genome.
ATTAAAGGTTTATACCTTCCCAGGTAACA1- (deletion at beginning)
C241T (substitution at 241)
C3037T (substitution at 3037)
A23403G (substitution at 23403)
TGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29867- (deletion at end)
Given these, it is impossible that Patient 3 caught Covid-19 from Patient 1.
Relative to the standard Covid-19 sequence, the virus from Patient 1 has lost (from its beginning) the segment
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT
However, the virus from Patient 3 has lost (from its beginning) only the shorter segment
ATTAAAGGTTTATACCTTCCCAGGTAACA
Also, the virus from Patient 1 has lost (from its end) the segment
CCATGTGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
However, the virus from Patient 3 has lost (from its end) only the shorter segment
TGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
Therefore Patient 1 cannot have transmitted the virus to Patient 3, unless the virus from Patient 1 somehow found the missing segment AACCAACCAACTTTCGATCTCTTGT, and attached it to its beginning, and also found the missing segment CCATGTGATTTTAATAGCTTCTTAGGAGAA, and attached it to its end, with these attachments occurring sometime during the transmission. But this is clearly impossible.
For the same reason, the claimed transmissions from Patient 4 to Patient 5, and from Patient 5 to Patient 7, are impossible.
So, what really did happen?
The standard Covid-19 sequence is called Wuhan-Hu-1/2019 or NC_045512.2 or MN908947.3 or EPI_ISL_402125.
The GenBank ids and mutations of the Munich cluster can be found here.
Most of the mutations from the Munich cluster occur in the data many times. The exceptions being G6446A and C22323T. These being the two new mutations, one discovered in Patient 4, and the other in Patient 9. Not including those from the cluster, we have the following numbers for the mutations of interest:
| 10247 | C241T |
| 10272 | C3037T |
| 1031700 | A23403G |
| 1 | G6446A |
| 6 | C22323T |
| 1279 | ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- |
| 52 | ATTAAAGGTTTATACCTTCCCAGGTAACA1- |
| 409 | TGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29867- |
| 159 | AATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29865- |
| 838 | CCATGTGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29837- |
A virus is considered a direct descendant of Patient 1 if it carries all the mutations of the virus of Patient 1, that is, its sequence contains, at least, the five mutations ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- C241T C3037T A23403G and CCATGTGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29837-.
I checked Patient 1 against a database (of 14,712 complete sequences), and found that Patient 1 has 1,050 direct descendants, with 728 of these being distinct, and these carried 958 different mutations.
The 1,050 direct descendants of Patient 1, and their mutations, are listed here.
Surprisingly, the article claims that the Munich cluster was contained, i.e., the virus was stopped from spreading further, it was (locally) wiped out. If true, this would mean that at some other place another virus would have had to mutate (from the Wuhan virus) in exactly the same way as the virus of Patient 1, that is, it would have to gain exactly the same five mutations of Patient 1, in order to explain the 1,050 descendant sequences. That such an amazingly unlikely event has to occur, tells you that the official story is, at least in part, wrong.
Except for seven sequences from the Munich cluster, and two from the USA, all the descendant sequences of Patient 1 are also members of the much larger British-American grouping whose sequences contain the four mutations C241T, C3037T, C14408T and A23403G. This group comprises 10,120 sequences of the 14,712 sequences for which I have data. This predominant group is mainly found in Europe, and North America. It is particularly rare in Asia, with only nine such sequences from China. Explaining how this grouping came about is so difficult that no one broaches the subject.
With nearly 70% of the Covid-19 sequences being of the British-American strain, the evolutionary tree should clearly be rooted at one of the sequences which contain only the four mutations C241T C3037T C14408T and A23403G. Sequences with only these four mutations are predominantly British, with 30 (of 72 in all) being from Britain.
Getting back to the Munich cluster, it seems probable that the Chinese woman, Patient 0, had the Wuhan strain, but did not spread it while in Germany. The others in the cluster caught the British-American strain that was spreading across Europe and North America. However, there is also the possibility that Patient 0 caught the disease on return to China, or even caught the British-American strain of Covid-19 while in Germany, and took it back to China. [The British-American strain is very rare in China with only 9 such sequences found. Descendant sequences of Patient 1 occurring in China are, of course, even rarer with only 3 sequences found.]
The unaligned viral RNA data (in fasta (fast A) format) for the patients of this case is here.
The aligned viral RNA data (in fasta format) for the patients of this case is here.
In the investigation above, I missed a rather important detail. Patient 4 was the source of a G6446A mutation which was passed to Patient 5, and from there to Patients 6, 7, 8, 9, 10, 11, 14, and 16. But, transmission of the virus from Patient 4 to Patient 5 has been shown to have been impossible. So, the G6446A mutation was either made up, or Patient 5 spread (a British-American strain of) the virus to Patient 4, and the others. However, we read that;
"The possibility that patient 4 could have been infected by patient 5 was excluded by detailed sequence analysis: patient 4 had the novel G6446A virus detected in a throat swab and the original 6446G virus detected in her sputum, whereas patient 5 had a homogeneous virus population containing the novel G6446A substitution in the throat swab."
Thus the G6446A mutation was made up. And, what then of the rest of the paper? It is unlikely that the entire paper was concocted from thin air. It is more likely that the basic aspects of the case are correct, with a thin genetic fiction laid over them. It is very unlikely that the sequences have been completely fabricated, otherwise they would not contain the heads, and tails, that they do. Before, it was possible, though not likely, that the paper was an honest misreading of the facts, but the G6446A mutation shows that it is a deliberate lie.
The G6446A mutation is extremely rare with only one other case known. There are also three G6446T mutations. All four of these are from England. It is suspicious that the researchers found the virus in the process of mutating, not once, which in itself is suspicious, but twice (the G6446A and C22323T mutations). Of course, this would be much more likely if the patients were infected with a cocktail of viruses.
Another indication of fabrication is the C241T C3037T A23403G mutations carried by the viruses of all the patients. These being the British-American mutations with C14408T missing. Such combinations are very rare with only 34 sequences (not counting the 14 of the cluster itself) among the 14,712. Twenty of these sequences are from Belgium, a country conveniently close to the Munich cluster. However, in a more recent Covid-19 collection, 18 of the sequences from Belgium had mysteriously regrown a C14408T mutation. I say regrown, because it is clear that these 18 originally had a C14408T mutation that was removed.
It seems that these Belgian sequences were being setup for a transition from the Munich type to the British-American type. Thus we would have the Wuhan type transitioning to the Munich type transitioning to the British-American type. However, the tampering of the Belgian sequences was all in vain, as for some reason the attempt was abandoned. Given the Belgian sequences, and the absence of any further sequences of this type in Germany, it seems likely that the Munich sequences were also British-American sequences with their C14408T mutation removed.
Anyway, adjusting for the tampering, the geographical distribution of sequences with, at least, the C241T C3037T A23403G mutations, but without C14408T, is Belgium 20 - 18 = 2, Germany 14 - 14 = 0, Spain 3, China 3, USA 2, Taiwan 1, Sweden 1, India 1, Greece 1, Denmark 1, and Bangladesh 1, a total of 16.
[The 18 Belgian sequences that were altered are: EPI_ISL_420345 EPI_ISL_420346 EPI_ISL_420347 EPI_ISL_420348 EPI_ISL_420349 EPI_ISL_420350 EPI_ISL_420351 EPI_ISL_420352 EPI_ISL_420353 EPI_ISL_420354 EPI_ISL_420355 EPI_ISL_420358 EPI_ISL_420359 EPI_ISL_420360 EPI_ISL_420361 EPI_ISL_420362 EPI_ISL_420363 EPI_ISL_420364 EPI_ISL_420365 EPI_ISL_420366]
The geographical distributions of other possible ancestor sequences are:
C241T C3037T C14408T without A23403G has the distribution, USA 7, Saudi Arabia 1, Netherlands 1, and Australia 1.
C241T C14408T A23403G without C3037T has the distribution, USA 30, Austria 10, Saudi Arabia 2, France 2, Wales 1, Sweden 1, Iceland 1, and England 1.
C3037T C14408T A23403G without C241T has the distribution, USA 27, Turkey 19, Jordan 13, Russia 12, Australia 5, Saudi Arabia 3, Brazil 3, Israel 2, France 2, China 2, and Scotland 1.
None of these provide a believable path from the Wuhan strain to the British-American strain, which indicates that the British-American strain was a separate release of the virus. Was it released before or after the Wuhan strain. Iceland seems to imply before, but Iran, and Italy, suggest after.
This file lists all 14,712 sequences of the database, and their mutations. These 14,712 sequences contain 8,047 distinct mutations.
More tampering with the data?
It seems that the fabricators of the Lancet paper also planted the following three sequences in GISAID:
EPI_ISL_416327 2020-01-28 hCoV-19/Shanghai/SH0014/2020 ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- C241T C3037T A23403G TGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29867-
EPI_ISL_416386 2020-01-31 hCoV-19/Shanghai/SH0086/2020 ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- C241T T1216C C3037T C6091T A16205G C17977A A23403G TGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29867-
EPI_ISL_416334 2020-02-06 hCoV-19/Shanghai/SH0025/2020 ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- C241T C1555A C3037T A9238G A23403G TGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29867-
The first two sequences have the same internal mutations (C241T C3037T A23403G) and same tail mutation (TGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29867-) as Patient 1, and have that same head mutation (ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1-) as Patient 3. The third sequence is the same with a couple of extra internal mutations.
All three sequences are from Shanghai, where Patient 0 was diagnosed with Covid-19, and all have dates close to the collection date of Patient 0's sample (which was supposedly Jan. 26).
These sequences were fabricated to help convince you that the completely false Lancet paper, is completely true.
Sequences containing C241T C3037T A23403G (but not C14408T) are rare everywhere, but extremely rare in China. The only other such sequences found in, or fabricated for, China, are:
EPI_ISL_451345 2020-01-24 hCoV-19/China/SC-PHCC1-022/2020 C241T C3037T A23403G
EPI_ISL_429080 2020-02-05 Guangzhou/GZMU0019/2020 C241T C3037T A23403G AAAAAAAAAAAA29892-
The paper has other indications of being fabricated.
It claims that "No specimens were available for sequencing for patients 12, 13, and 15," and that "Sequencing of patient 15 was not successful." These being contradictory claims. And since the sequence for patient 15 was submitted under the name SARS-CoV-2/human/DEU/BavPat15-ChVir1484-ChVir1536/2020 [EPI_ISL_450211], both of these claims appear to be false.
Patient 12 was supposedly the first Covid-19 case to be diagnosed in Spain. It is surprising that his virus was not sequenced. Perhaps the Spanish authorities believed this had already been done. It turns out that this was not the case as the Lancet paper states "After Spanish authorities were informed, patient 12 was isolated in hospital on Jan 30 and diagnosed with Covid-19." So the mystery remains. This may mean that the story of the Munich cluster was fabricated at a later date, and was retrospectively "made true". However, this would involve creating false news reports, etc, and placing them with false dates. Not impossible if you are a large well organized group, as the spreaders of the virus seem to be.
Of course, this completely false paper, and its spinoff, were among the main drivers for the belief in asymptomatic transmission. Although many studies (regarding asymptomatic transmission) have been done, the CDC (Centers for Disease Control and Prevention, July 10, 2020) could only say, "The relative infectiousness of asymptomatic cases to symptomatic cases remains highly uncertain as asymptomatic cases are difficult to identify and transmission is difficult to observe and quantify. The estimates for relative infectiousness are (only) assumptions based on studies of viral shedding dynamics."
Here are a couple of lines from two review articles on Asymptomatic Transmission.
"This review identified and summarised 18 case studies reporting pre-symptomatic or asymptomatic transmission. However, their level of evidence is low and is subject to a number of potential sources of bias and therefore they should be interpreted with caution.... Based on the totality of the evidence presented in this report,it seems likely that pre-symptomatic transmission is occurring. Evidence of asymptomatic transmission from asymptomatic carriers, is more limited (perhaps due to difficulties in identifying truly asymptomatic carriers); it appears plausible, but it may not be a driver of transmission."
https://www.hiqa.ie/sites/default/files ... VID-19.pdf
"Our study does not support claims that the majority of SARS-CoV-2 infections are asymptomatic. Questions remain as to the role of asymptomatic carriers in the transmission of SARS-CoV-2."
https://www.medrxiv.org/content/10.1101 ... 1.full.pdf
On another reading of the paper I found the statement "The directly observed substitution rate was two substitutions per 29903 nucleotides per 11 days, equalling 2.2×10^-3 substitutions per site, per year."
0.0022 = 2.2×10^-3 = (2/29903)*(365/11) subs/site/year [or 66 subs/year]
The fact that the mutations were on different descendant viruses cleared up the question of which mutations are counted in the calculation of the substitution rate. All mutations of descendant viruses are counted. I checked Patient 1 against a Covid-19 database (of 14,712 complete sequences), and found that Patient 1 has 1,050 direct descendants, with 728 of these being distinct, and these carried 958 different mutations. We also have that these mutations occurred between Jan 24 and mid May, over a period of say, 120 days. Thus a rough estimate of the evolutionary substitution rate for this branch can be calculated.
It is (958/29903)*(365/120) = 0.097 = 9.7×10^-2 subs/site/year [or 2,914 subs/year]
This is impossibly high. Not only is the substitution rate impossibly high, but the direct descendants spread around the world in an impossibly short time (especially since at the beginning of the period airlines had considerably reduced their services). The geographical distribution of the direct descendants is Australia 1, Belgium 36, Brazil 1, Canada 16, China 3, Colombia 54, England 22, Scotland 165, Wales 2, Germany 7, Hungary 3, Ireland 7, Japan 14, Netherlands 209, Portugal 24, Serbia 2, Spain 39, Sweden 1, Thailand 2, and USA 442.
Another "peer-reviewed" paper designed to mislead.
The paper, Clinical and virological data of the first cases of Covid-19 in Europe: a case series, from "The Lancet Infectious Diseases," also misleads the reader concerning RNA data. Similar to the last, it has a large number of authors. One guesses that the twenty-three authors are to deflect blame, so that blame cannot be pinned on any one particular author, and thus ultimately, on none of them. With the same style and intent, it appears that both of these papers were ghost written by the same people.
Among the more important pieces of data to be gathered about these first cases are the viral RNA sequences. They were indeed gathered, and here is what the paper has to say about them:
"When available, the sequence analysis of the virus of these patients showed that patients 1 and 4 compared with patient 5 correspond to two distinct events of importation. For patients 1 and 4, the virus was clustering with viruses from cases in Wuhan, Shenzhen (China), California (USA), Australia, and Taiwan, whereas for patient 5 the virus was clustering with those from Chongqing (China) and Singapore (the genetic epidemiology of SARS-CoV-2 is available online). Furthermore, the very high degree of identity of the sequences from patients 1 and 4 supports the epidemiological link between these cases and the likelihood of transmission."
From this it is clear that the data is not to their liking, so they tell you essentially nothing, give you no links to the data, and tell you to go find it on the internet, if you can. Disgusting.
The deliberate spread of the virus to a New Zealand nursing home.
While looking for evidence that the British-American strain is not that dangerous, I came across the case of the Rosewood Rest Home, located in Christchurch, New Zealand, from which 12 people died. [It turns out that the British-American strain is just as dangerous as the other strains, if not more so.]
The relevant sequences and their mutations can be found here.
It appears that most of those who died carried the six mutations C241T, C3037T, C4683T, C14408T, A23403G, and G24077T. That is, the British-American mutations plus C4683T, and G24077T. As a group C241T, C3037T, C4683T, C14408T, A23403G, and G24077T are only found in 9 sequences from (Christchurch) New Zealand.
By itself, the mutation, C4683T, has the following geographical distribution:
New Zealand 9 sequences, United States 5, China 2, Singapore 1, and Australia 1.
By itself, G24077T, has the following distribution:
Portugal 65, United Kingdom 14, New Zealand 13, Netherlands 7, Iceland 3, United States 1, Switzerland 1, Italy 1, Georgia 1, Estonia 1, and Austria 1.
There are no possible ancestor sequences containing C4683T. Well, none if you discount such unlikely possibilities as the two Chinese viruses, just mentioned above, which would have to lose two specific mutations, and gain five extra, all at once. About the same probability as being able to hold your breath free-diving to the bottom of the Mariana Trench.
All sequences containing G24077T also contain the British-American mutations, so C241T C3037T C14408T A23403G G24077T has the same geographical distribution as G24077T. Apart from one sequence from the United States they are all European. How an ancestral sequence got to New Zealand, and then immediately mutated to the Christchurch form, is a bit of a mystery.
Overall, it looks like a nasty strain of the disease was deliberately released in the nursing home to affect New Zealand Covid-19 policy. What is particularly suspect about all this, is that the New Zealand Health authorities felt that there was nothing to report here. There were no questions as to where the virus came from. There were no questions as to whether these unusual mutations could have made the virus more lethal. There was no mention of the fact that this deadly viral form was unique to New Zealand, etc, etc. It's as if they went to the trouble of sequencing the virus, then threw away the results without looking at them.
Coverup down-under.
During April, and May, 2020, the Western Sydney nursing home, Newmarch House, suffered a deadly outbreak of Covid-19. The Australian current affairs program, Four Corners, investigated the outbreak, and produced a documentary called Like the plague.
The documentary points out that Newmarch House patients were not transfered to hospitals. Why?
One reads that "NSW Health is running the testing at Newmarch House while the federal government has provided an infection control specialist (Aspen Medical) who is monitoring and reviewing all current contamination and infection control procedures at the home."
Hospitals were emptied to take the predicted hundreds of thousands of sick and dying. But when a few people from Newmarch House needed to be transfered to one of these hospitals, health officials (one guesses on the advice of Aspen Medical) refused to allow this. This is truly incomprehensible.
On May 4, 2020, the Sydney Morning Herald carried this report;
New South Wales Health authorities are investigating whether multiple sources of infection caused the devastating outbreak of Covid-19 at the Anglicare Newmarch aged care facility. The investigation comes as a 15th resident at (another) nursing home in Caddens in western Sydney died from the virus on Monday, and amid growing concern among families of residents about the facility's ability to contain the outbreak. New South Wales Chief Health Officer Dr Kerry Chant said a forensic investigation was still ongoing into how coronavirus entered the nursing home, saying it was possible a previously identified employee was not the initial source of the outbreak. "What we're trying to look at is were there any other introductions into the aged care facility at or about that time," Dr Chant said. "Was she [the employee] the first case, or had she in fact acquired it from someone else?" "We know that the [completed] genomic sequencing is showing that the virus that spread into the nursing home is a virus that was circulating in the community, there's linkages between a number of other clusters," Dr Chant said. "This is forensic detective work. We are just trying to look at whether there are missing links or missing chains."
The relevant viruses were sequenced, and detective work was conducted to map the spread of the virus. As far as I am aware, the results of this work were never made public. In fact, it appears that a great effort was made to hide away the data. The RNA sequences were almost certainly submitted to GISAID. However, GISAID has not released any Covid-19 sequence submitted from Sydney since April 19 2020. Not even one. You should contrast this with viral information from non-Sydney, New South Wales, locations. GISAID has released every non-Sydney sequence that has been submitted to them, every single one, even the ones submitted to them on July 29 2020.
With the first Covid-19 case identified at Newmarch House on 10 April, and given 4 or 5 days to get the sequencing, etc, done, the timing of the GISAID black-out is rather suspicious. Chant mentions the investigation showed linkages between clusters. It seems likely that the detective work did indeed provide a link between various disease clusters, a link that indicated that the virus was being deliberately spread.
I have recently acquired a collection of 40,132 Covid-19 sequences (and now 77,827). In this file I list all the Covid-19 sequences submitted to GISAID from New South Wales, that is, all the Covid-19 sequences from New South Wales in their database. For sequences in the collection I have also added the mutations carried by the sequences. Sydney sequences are labeled "Australia/NSW/Sydney". Sequences from outside Sydney are labeled simply "Australia/NSW". "No Data" indicates that the sequence has been submitted, and thus in their database, but not in the collection.
In order to investigate this further I have acquired another collection. This one has 77,827 Covid-19 sequences.
This file lists the 77,827 sequences and their mutations in .cvs format.
This file lists the 77,827 sequences, as just above, but with more detailed information concerning the location.
These 77,827 sequences contain 23,692 distinct mutations.
Unfortunately, the new collection did not provide any of the missing NSW sequences, not even one.
And, just in case you think it is only GISAID, and Nextstrain, that are hiding/fudging the facts, I present the number of sequences from Australia that are among the 15,217 currently (Aug. 9 2020) being distributed by NCBI;
Australia: Victoria 1379
Australia: Northern Territory 31
Australia: South Australia 4
Australia 4
Australia: Queensland 2
Note that the number of sequences from New South Wales, Australia, is zero (the 4 sequences labelled simply Australia are from Queensland). Given Sydney is the most populous city, and has had significant Covid-19 outbreaks where we know viruses were sequenced, there is a suspicious lack of New South Wales sequences. NCBI stands for the National Center for Biotechnology Information, and is a United States company.
To further investigate this I downloaded the 23,529 sequences currently (Sep. 23 2020) available from NCBI. The number of sequences from New South Wales, Australia, is still zero. Here are the current statistics for Australia:
Australia: Victoria 5317
Australia: Northern Territory 34
Australia: South Australia 4
Australia: Tasmania 200
This 8.8M gzipped file contains the 23,529 sequences in fasta format.
The uncompressed file from NCBI is 687M. It makes you wonder why they do not compress their files.
The details, including mutations, of this collection are listed here in .csv format.
One notes that Sydney has 27 sequences with the following unusual head and tail mutations:
ATTAAAGGTTTATACCTTCC---226-bases-missing---GGTGTGACCGAAAGGTAAGA1-
CAATCTTTAATCAGTGTGTA---189-bases-missing---AAAAAAAAAAAAAAAAAAAA29675-
I will call them ATTAA*TAAGA1- and CAATC*AAAAA29675- for short.
Sequences carrying (at least) these two mutations are not found anywhere else in Australia. The geographical distribution of such sequences is United States 780, Australia 27, Mexico 25, Jordan 23, Ecuador 5, Singapore 1, a total of 861. I hope I do not have to point out that China is not on this list.
The 861 include whole groups of known sequences with their ends severely trimmed. Of course, this is all very, very strange. Such a collection of sequences could not develop naturally. It is like someone took a pair of scissors to a large number of existing sequences, and trimmed the ends of all of them at exactly the same two points. This was so unusual that I had to take a closer look.
I will call the 861 sequences having the CAATC*AAAAA29675- mutation, simply 29675-sequences.
I looked for ancestor sequences, and found that nearly all 29675-sequences have an ancestor sequence that was collected on the same day, and at the same place, as the 29675-sequence. In total I found 745 [29675, ancestor] pairs, with both members of each pair having the same collection date, and location. This is obviously not from a natural occurrence. So, which are fake, the 29675-sequences, or the ancestor sequences associated to them?
Since 29675-sequences have been found by at least three different laboratories, it seemed likely that the 29675-sequences were not simply the result of some strange glitch in the sequencing process. The three laboratories being Westmead Hospital, University of Sydney, Australia [27 sequences], the Instituto Nacional de Investigación en Salud Pública, Ecuador [5 sequences], and Duke-NUS Medical School, USA [1 sequence]. I said "at least three" because the entry providing the originating lab has been deleted from 827 of the 861 records, so there may well have been many more than three.
Almost all the records with the originating lab entry deleted have "The Scripps Research Institute, USA" as the submitting lab. And, what of the ancestor component of the [29675, ancestor] pairs? Well, 744 of the 745 ancestor sequences list "The Scripps Research Institute, USA" as both the originating lab, and submitting lab. Maybe the Scripps Research Institute should be asked a few questions.
Adding a near full length sequence extending each of the 29675-sequences is a way of hiding the 29675-sequences, whereas faking the 29675-sequences gains nothing.
From all this it is clear that the ancestor component of each of the pairs is entirely fabricated.
You can view the 745 [29675, ancestor] pairs here. They have the four line format:
ID of 29675-genome; Collection date of sample; Length of genome; Name of genome; Location;
Mutations of 29675-genome (common mutations in red)
Mutations of associated ancestor genome (common mutations in red)
ID of ancestor genome; Collection date; Length of genome; Name of genome; Location;
An example being:
MT810526 2020-03-20 29409 SARS-CoV-2/human/USA/SEARCH-1660-SAN/2020 United States/San Diego/California
| ATTAAAGGTT---265-bases-total---GAAAGGTAAG1- | C3037T C14408T A20268G A23403G CAATCTTTAA---229-bases-total---AAAAAAAAAA29675- |
| ATTAAAGGTTTATACCT1- C241T | C3037T C14408T A20268G A23403G |
Note that both the 29675-sequence, here MT810526, and the ancestor sequence, here EPI_ISL_494721, invariably have the same collection date, and location. Note also that the 29675-sequence location is invariably written country/city/state while the ancestor sequence is written country/state/city, and that the same number, in this case 1660, is invariably used in both the 29675 and ancestor sequence names.
So, how did the 29675-sequences originate? That is a hard one to answer. Over four hundred different existing viruses have had their ends trimmed to give the 861 sequences of 29675-type. It seems that many different viruses must have been found together in one place, when their ends were cut off at exactly the same two points (relative to the standard sequence). Perhaps an RNA cutting/editing protein was added in order to change one or more of the viruses in some desired way, and this protein was accidently left in a number of vials, each of which contained a cocktail of covid-19 viruses. Of course, such an editing protein should have been thoroughly removed from the mixture. However, in some cases it appears that it was not successfully purged, and in time, it amputated the ends of each covid-19 virus in the vial.
The 29675-sequences are closely related to outbreaks on cruise ships. Two cruise ships with 29675-sequences docked at San Diego. One of these was the Disney Wonder. The Disney Wonder had previously stopped in at New Orleans, Louisiana. Both San Diego and New Orleans had an outbreak of viruses of the 29675-type. Of course, Sydney Australia also had a 29675-type outbreak. One assumes this was the type of the Newmarch House outbreak, and this led to the above mentioned cover-up. Newmarch House also had a connection to cruise ships, as Aspen Medical staff who had worked on the cruise ship Ruby Princess later worked at Newmarch. The 25 sequences of 29675-type from Mexico are all from Tijuana which neighbors San Diego. The 23 sequences of 29675-type from Amman, Jordan, probably have a connection to the US military.
Since the latest collection (originally from GISAID) did not provide any of the missing NSW sequences, and some of the collection's data appears to have been tampered with (e.g., the dates of certain Spanish sequences), I started looking for older collections. And, the very first one, from June 27, provided 393 of the missing NSW sequences. I had guessed that what was being hidden was a bunch of 29675-sequences, and 229 of them were, but there were also 164, 29677-sequences.
A 29677-sequence is what you would expect, i.e., a sequence with the tail deletion
ATCTTTAATCAGTGTGTAACATTAGGGAGGACTTGAAAGAGCCACCACAT
TTTCACCGAGGCCACGCGGAGTACGATCGAGTGTACAGTGAACAATGCTA
GGGAGAGCTGCCTATATGGAAGAGCCCTAATGTGTAAAATTAATTTTAGT
AGTGCTATCCCCATGTGATTTTAATAGCTTCTTAGGAGAATGACAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAA29677-.
It is usually paired with the head deletion
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTC
TTGTAGATCTGTTCTCTAAACGAACTTTAAAATCTGTGTGGCTGTCACTC
GGCTGCATGCTTAGTGCACTCACGCAGTATAATTAATAACTAATTACTGT
CGTTGACAGGACACGAGTAACTCGTCTATCTTCTGCAGGCTGCTTACGGT
TTCGTCCGTGTTGCAGCCGATCATCAGCACATCTAGGTTTCGTCCGGGTG
TGACCGAAAGGTAA1-
I will call these two mutations ATCTT*AAAAA29677- and ATTAA*GGTAA1- for short.
The 393 new sequences, and their mutations, can be found here.
The old plus new sequences together are listed here.
The first 29675-sequence was collected on Feb. 3 in Singapore.
The second 29675-sequence was collected on Feb. 28 in Sydney.
The first 29677-sequence was collected on Feb. 28 in Sydney.
Following the natural order of things the 29677-sequences should have occurred first.
Everything is weird. One guesses that these Sydney sequences had to be part of the same Covid-19 cocktail.
All 29677-sequences are from Sydney, Australia, except one from Spain, and one from Portugal:
EPI_ISL_454103 2020-03-20 2020-05-28 hCoV-19/Portugal/PT0387/2020 Portugal
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAAC1- C241T C3037T C14408T A23403G C26408T GGG28881AAC ATCTT*AAAAA29677-
EPI_ISL_455336 2020-02-29 2020-05-29 hCoV-19/Spain/LaRioja201277/2020 Spain/LaRioja
C8782T T9477A C14805T G24040T G25979T T28144C C28657T C28863T ATCTT*AAAAA29677-
Such a distribution is hard to explain.
A strange feature is that three or four days after the number of 29677-viral-sequences peaked, they disappeared from the face of the planet, never to be seen again. The 29677-sequences (just like the 29675-sequences) are actually amputated versions of many different genomes. In fact, the 29677-sequences are amputated versions of 115 different preexisting genomes. So, how is it that all 115 different amputated genomes choose to disappear at exactly the same time, and just after their peak occurrence? The answer is obvious. They all disappeared when the viral cocktails from particular vials were no longer spread.
Here is a graph recording the numbers of 29675- and 29677-sequences collected each day between Feb. 23 and May 20.
Another tail mutation, CCATG*AAAAA29837- for short, is extremely common. A virus with the CCATG*AAAAA29837- tail mutation will be called simply a 29837-virus, and a sequence with such will be called a 29837-sequence. There are 25,303 such 29837-sequences in the 77,827-collection. 24,711 of these are paired with the head mutation ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1-. The 29837-viruses (just like the 29675-viruses and 29677-viruses) are descendants of amputated versions of many different preexisting genomes. In fact, the 29837-viruses are descendants of the amputated versions of 672 different preexisting viruses/genomes.
It is notable that in the earlier 14,712-collection there are zero 29837-sequences, zero 29675-sequences, and zero 29677-sequences.
Overall, the main variants of the virus appear to be only mildly contagious, so to keep the pandemic going the virus has to be actively spread.
It seems that initially aircraft and cruise ships were used to spread the virus. Old-age homes were also targeted.
This explains why China recovered so quickly from her dose of the virus. Although the virus had been actively spread in Wuhan, leading to reports of the virus being very contagious, the lockdown, and the lack of old-age homes, kept the spreaders from spreading it further. Thus they moved on to easier, or more hated, targets. Western countries proved particularly soft targets as the disease could be spread to the numerous old-age homes. Such old-age homes are rare in Asia as the older population mostly lives with their children. Since the virus is only mildly contagious, the lockdown soon killed off the little that had previously spread to the rest of China. It seems the spreaders expected the virus to be much more contagious than it turned out to be, as they spread it just before the biggest mass movement of people anywhere on the planet, Chinese new year.
Cruise ships used to spread the virus.
5 April 2020: New South Wales Police Commissioner Mick Fuller announces a criminal investigation into the operator of the Ruby Princess cruise ship after the death of 10 passengers from coronavirus. This was to be conducted by the NSW Police homicide squad.
15 April 2020: A Special Commission of Inquiry into the Ruby Princess was initiated to curtail the police investigation. Some months later the Commissioner reported that everything was an unfortunate string of accidents and errors. The idea here is to use the report of the special Commission as an excuse to wind down, and quietly drop, the police investigation. However, this will probably not stop the group of 800 passengers who are pursuing a class action lawsuit. (Those pursuing this class action should be very wary of their leaders, as it is unlikely these people are, in reality, on their side.)
22 April 2020: NZ Customs announced an investigation into Ruby Princess cruise-ship staff concerning Covid-19 clusters in New Zealand. It seems that this "investigation" has also been quietly dropped.
Accidental, or not, the Ruby Princess certainly spread the virus far and wide:
Here is a false declaration of health sent from the Ruby Princess to the Auckland District Health Board. It was obtained by stuff.co.nz under the Official Information Act. The declaration is dated 16 March, and claims to have been submitted at the port of Tauranga, however, the cruise of the Ruby Princess was cut short, and the ship never visited Tauranga.
On 15 March a declaration of health would have been sent from the Ruby Princess, then at the port of Napier, to the Bay of Plenty District Health Board. This would have been needed to dock at the port of Tauranga, as planned for March 16. After changing one word, Napier, to Tauranga, and the (first instance of the) date to the 16th, and presumably the registration number, the same document was sent to the Auckland District Health Board. This would have been needed to dock at Auckland, as planned for March 17. Doing this had the effect of hiding the true health situation on March 16. Why was it necessary to hide the true health situation of the Ruby Princess? One has to assume that someone on board the ship was aware that it was carrying Covid-19.
And another paper designed to mislead.
While searching for a relation between mutations, and death rate, I came across this article.
The article reads very strangely, and it took some hours to decipher. First, they mention nine very important mutations, but never tell you what they are. However, you can find them labeling diagrams at the end of the article. They are C8782T, C17747T, A17858G, C18060T, T28144C, C241T, C3037T, C14408T, and A23403G. The last four you are already familiar with, they are the four mutations of the British-American strain. They mention that C241T, C3037T, C14408T, and A23403G are mainly European, and exist as a group, as you already know.
Then they mention that the mutations C17747T, A17858G, and C18060T are mainly American, and are nearly always found together as a group. This was intriguing as I had entirely missed this grouping. I wondered about the other two mutations C8782T, and T28144C. It didn't take long to find out that these two plus C17747T, A17858G, and C18060T form a tight group of five mutations. So, why did the authors report only a group of three?
It eventually dawned on me that the reason for the overall obscurity of the article, and the misdirection, is to prevent the reader discovering the group of five. This is because the group of five is impossible to explain as a natural consequence of the official Covid-19 tale. So what do we know about the group of five?
The group of five [C8782T, C17747T, A17858G, C18060T, and T28144C] occur in 1146 sequences.
To gain some idea of just how tight this group of five is, consider the following:
Sequences containing C8782T, C17747T, A17858G, and C18060T, without T28144C = 3.
Sequences containing C8782T, C17747T, A17858G, and T28144C, without C18060T = 5.
Sequences containing C8782T, C17747T, C18060T, and T28144C, without A17858G = 0.
Sequences containing C8782T, A17858G, C18060T, and T28144C, without C17747T = 19.
Sequences containing A17858G, C17747T, A17858G, and C18060T, without C8782T = 3.
There are 164 sequences which carry only these five mutations (and no more). They are entirely from North America, with 160 from the USA and 4 from Canada. Hence, the evolutionary tree of the American strain is clearly rooted in North America, that is, this strain started in North America, and spread elsewhere.
Descendant sequences, that is, sequences containing at least the group of five mutations are predominantly American, with 1042 (of 1146 in all) from the USA. The distribution is: USA 1042, Australia 49, Canada 26, Costa Rica 1, England 2, Iceland 15, Mexico 4, Puerto Rico 3, Taiwan 3, and Uruguay 1.
Explaining how the group of five developed from Wuhan is probably impossible, but anyway it is so hard to explain that explanations are not offered, and the topic is hidden away.
As to the correlation of mutations to death rate, the authors state that they have removed from consideration all sequences which are the same as the standard Wuhan genome. This invalidates their results. The 2nd edition says nothing of what the correlations actually represent. In the 1st edition one has;
"The frequencies of specific sites for each country were calculated. The death rate was estimated with Total Deaths/Confirmed Cases based on the data from Johns Hopkins resources on April 22th, 2020. The correlation coefficient between death rate and frequencies of specific site or haplotype in different countries was calculated using Pearson method."
With this as a strategy, I can see why they deleted this sentence in the 2nd edition. Better to say nothing.
More viruses mysteriously regrow their tails.
For the period 2020-01-19 until 2020-02-20 (the first month of the America Covid-19 epidemic) every single American viral sequence had lost AAAAAAAAAAAAAAAAAAAAA, or more, from it's tail. In particular, all potential ancestors of the group of five had AAAAAAAAAAAAAAAAAAAAA, or more, missing from their tail. Here is a list of all the potential ancestors of the group of five from that first month:
2020-01-19 MN985325 C8782T C18060T T28144C AAAAAAAAAAAAAAAAAAAAA29883-
2020-01-19 MT233526 ATTAAAGGTTTATACCTT1- C8782T C18060T T28144C ATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29866-
2020-01-19 MT246667 ATTA1- C8782T C18060T T28144C AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29871G
2020-01-22 MN997409 C8782T G11083T T28144C C29095T AAAAAAAAAAAAAAAAAAAAA29883-
2020-01-23 MN994467 G1548A C8782T C24034T T26729C G28077C T28144C A28792T AAAAAAAAAAAAAAAAAAAAA29883-
2020-01-25 EPI_ISL_407214 C8782T C18060T T28144C AAAAAAAAAAAAAAAAAAAAA29883-
2020-01-25 EPI_ISL_407215 C8782T C18060T T28144C AAAAAAAAAAAAAAAAAAAAA29883-
2020-01-28 EPI_ISL_410045 T490A C3177T C8782T C24034T T26729C G28077C T28144C AAAAAAAAAAAAAAAAAAAAA29883-
2020-02-06 EPI_ISL_411954 C8782T T28144C G28878A G29742A AAAAAAAAAAAAAAAAAAAAA29883-
2020-02-11 EPI_ISL_411956 C8782T T18603C T18975A A19175C C27925T T28144C C29095T AAAAAAAAAAAAAAAAAAAAA29883-
Then on 2020-02-20 something strange happened. A sequence turned up with a full tail. The first one. It's id was EPI_ISL_413456. It also carried the group of five mutations. Then a few more a couple of days later. Then there were 889 sequences carrying the group of five mutations and having a full tail. How does a true believer explain this? I guess the only possible explanation is for the group of five, plus full tail, to have been introduced to North America from outside, sometime around 2020-02-20, when the first case magically appears. But it turns out that there is nowhere (from outside North America) it could have come from. Here is a full list of possible ancestors countries and mutations:
111110 USA 510 Australia 9 Canada 9 England 2 Iceland 14 Taiwan 2 Uruguay 1
111100 USA 2
011110 USA 2
101110 USA 1 Australia 1 Canada 13
111010 Iceland 1
100110 USA 2
100010 USA 91 Australia 15 Canada 1 China 14 Denmark 1 England 4 France 1 Germany 1 Japan 1 Jordan 1 Kazakhstan 1 New Zealand 2 Russia 2 Saudi Arabia 12 Scotland 15 Senegal 2 United Arab Emirates 1 Unknown 1
100000 Scotland 1 Wales 1
010000 Wales 1
000100 USA 1 England 1
000010 USA 1 South Korea 1
000000 All countries 6235
The mutations are represented by the initial binary number. For example, 100010 is shorthand for 1 mutation C8782T, 0 mutations C17747T, 0 mutations A17858G, 0 mutations C18060T, 1 mutation T28144C, and 0 mutations at the end (i.e., a full tail). The unlisted binary numbers. e.g., 110110, represent combinations of the mutations that have not been found anywhere in the world. Clearly, all ancestor sequences must have a full tail, i.e., the last digit of the binary number must be zero.
I guess, if one is really desperate, one has to look at the 14 Chinese sequences among those labeled 100010. This is a real, real long shot, for such ancestor sequences need to gain the three missing mutations, C17747T, A17858G, and C18060T, and at essentially the same time. To make matters even worse, 13 of the 14 sequences carry other mutations which make it impossible for them to be ancestors. So, one is left with EPI_ISL_413858, from Guangdong province, which carries only the mutations C8782T, and T28144C (and has a full tail). Even if you believe this extremely unlikely triple-jump happened, you are still left with a host of problems, like; How did the virus EPI_ISL_413858 get to America? And if it did, why did it spread in America but not in China? etc, etc.
When you are reduced to believing such incredibly unlikely events, the story you believe is probably wrong. Here, it is much, much easier to believe the group of five is native to North America, and independent of Wuhan.
Tight groupings in the data.
I adjusted the script used in the last section in order to find all significant groupings among the twenty most common mutations. This is what I found:
C1059T G25563T == First Group of Two (3624 of 14712 sequences carry these mutations)
C8782T T28144C == Second Group of Two (1807 sequences; includes the Group of Five)
G11083T C14805T G26144T == Group of Three (814)
C241T C3037T C14408T A23403G == Group of Four (10120)
C241T C3037T C14408T A23403G G25563T == Group of Four + G25563T (4329)
C241T C3037T C14408T A23403G C1059T == Group of Four + C1059T (3585)
C241T C3037T C14408T A23403G C1059T G25563T == Group of Four + First Group of Two (3564)
C241T C3037T C14408T A23403G GGG28881AAC == Group of Four + GGG28881AAC (2953)
C241T C3037T C14408T A23403G G25563T == Group of Four + AATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- (790)
C241T C3037T C14408T A23403G A20268G == Group of Four + A20268G (732)
C241T C3037T C14408T A23403G C18877T == Group of Four + C18877T (439)
C241T C3037T C14408T A23403G C1059T G25563T C27964T == Group of Four + First Group of Two + C27964T (359)
C8782T C17747T A17858G C18060T T28144C == Group of Five (1146) < Second Group of Two
These are the major groupings. The Group of Four+Two is a group among the descendants of the Group of Four, and the Groups Four+Two+X are groups among the descendants of the Group of Four+Two, etc. How these groupings developed is impossible to explain without throwing away the official explanation.
So you can judge which are tight groupings, the number of individual occurrences, in square brackets, are compared to the group occurrences, in parentheses.
C1059T [3654 sequences] G25563T [4426 sequences] (3624 sequences as a group)
C8782T [1835] T28144C [1816] (1807)
G11083T [1477] C14805T [999] G26144T [946] (814)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] (10120)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] G25563T [4426] (4329)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] C1059T [3654] (3585)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] C1059T [3654] G25563T [4426] (3564)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] GGG28881AAC [2994] (2953)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] AATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT1- [1285] (790)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] A20268G [745] (732)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] C18877T [469] (439)
C241T [10258] C3037T [10283] C14408T [10283] A23403G [10328] C1059T [3654] G25563T [4426] C27964T [384] (359)
C8782T [1835] C17747T [1158] A17858G [1177] C18060T [1188] T28144C [1816] (1146)
That C3037T and C14408T were both in exactly 10,283 sequences was surprising. I checked in the 40,132 collection and found C3037T was in 31,212 sequences and C14408T in 31,169. Together, they were in 31,040 sequences.
Yet another paper designed to mislead.
The preprint "Identification of novel missense mutations in a large number of recent SARS-CoV-2 genome sequences," is another example of a paper that was deliberately designed to point researchers in the wrong direction. First, I wondered about this statement:
"On April 11, 2020, there were 547 SARS-Cov-2 sequences deposited in GenBank, from which, we downloaded 474 complete or near-complete genomes..."
So, I downloaded the Covid-19 sequences available from GenBank (over 8000), removed duplicates, animal sequences, and sequences of length less than 29000. This left me with 7683 sequences. According to GenBank's own data, 1555 sequences had been submitted to them by April 11. So why were only 547 sequences available for download?
My guess is that GenBank deliberately held back the submitted data while giving certain people access to it so they could concoct a story that hid their nefarious activities. Once they had established the narrative, the raw data (perhaps slightly adjusted) could be released to the scientific public.
This process allows a certain degree of cherry-picking. If you cherry-pick 474 sequences from 1423, as they were likely able to do, then people will ask questions, but if 474 have been chosen from 547, then that sounds reasonable.
The mutations of this GenBank collection are listed here.
So, if there was cherry-picking, what was its purpose? Consider this statement:
"Our analysis highlights 5 frequent new mutations that have emerged since late February 2020. These mutations are: one each missense (non-synonymous) mutation in orf1ab (C1059T), orf3 (G25563T) and orf8 (C27964T), one in 5'UTR (C241T), one in a non-coding region (G29553A). The final mutation (G29553A) was found to be almost exclusive to the US isolates."
Our authors report the following five new mutations: C1059T G25563T C27964T C241T G29553A.
From the 1423 sequences they (probably) had available to them, we have
308 C1059T mutations (22% of the 1423)
335 G25563T 24%
34 C27964T 2%
65 G29553A 5%
499 C241T 35%
We also have the following mutations that the authors managed not to find. Each of them more prevalent than those they did find.
519 C3037T 36%
506 C14408T 36%
517 A23403G 36%
The percentages for earlier dates decrease proportionally to zero. For later dates they increase proportionally until each of C241T, C3037T, C14408T and A23403G is 65%.
As you know, from above, C241T, C3037T, C14408T and A23403G exist as a group. If you find the mutation C241T, as the authors did, then you also find the mutations C3037T, C14408T and A23403G. They are tied together. It is impossible to find the mutation C241T without finding the other three. Yet our authors managed to miss this very large elephant grazing in a very small china shop. So, hiding the British-American strain seems to be one object of the paper.
In fact, it is even worse than just stated. As you can see four (C1059T G25563T C27964T C241T) of the five mutations found by the authors are actually part of the Group of Four+Two+One (C241T C3037T C14408T A23403G C1059T G25563T C27964T). That is, four of the five mutations found by the authors are part of this tight grouping of seven. That is, four of the five mutations found by the authors are so closely tied to C3037T, C14408T and A23403G that it is literally impossible to miss them. Yet "miss" them they did.
Another peer-reviewed, but mostly wrong, paper.
The peer-reviewed paper "Phylogenetic network analysis of SARS-CoV-2 genomes," states: "Finally, to ensure comparability, we truncated the flanks of all (RNA viral) sequences to the consensus range 56 to 29,797, with nucleotide position numbering according to the Wuhan 1 reference sequence." Doing this invalidates their results.
So, the authors trimmed the RNA to some "consensus range" to ensure "comparability". That is, they deleted the information that was stopping them getting the results they wanted. The "consensus range" was the minimum trim necessary to be rid of all the undesired mutations, so the authors understood what they were doing. Maybe they figured that future sequences would justify this fudging of the facts.
To see if ignoring the lower and higher numbered mutations actually made any difference, I checked the twelve sequences (EPI_ISL_406036 EPI_ISL_410713 EPI_ISL_411951 EPI_ISL_412029 EPI_ISL_411929 EPI_ISL_406031 EPI_ISL_412116 EPI_ISL_410536 EPI_ISL_406844 EPI_ISL_406597 EPI_ISL_410714 EPI_ISL_410546) claimed to be descendant from the Sydney virus EPI_ISL_408977. None of these cases of paternity were supported by the facts (i.e., the full sequences). You can find the results here. I didn't check any further.
Another complaint is that the supplement shows the bat coronavirus RaTG13, GenBank id MN996532.1, is linked to the standard Covid-19 sequence by the mutations T28144C C8782T T29095C C18060A C3037T G4255T C6031T C7420T C10138T A11707G C12115T T15597C C17373T C18828T C21859T T22303C C24034T C25587T, and T25645C. This is wrong. The stated mutations are just a small sampling of the 1109 mutations by which the bat sequence and the standard sequence differ.
Is it possible that the bat virus BatCoVRaTG13 jumped species directly to the Wuhan strain? In a word, no. The two only have 96.2% sequence similarity, that is, there are 1189 base differences. These 1189 base differences make up the 1109 mutations mentioned above.
Note that the first two mutations mentioned in the paper are incorrectly named. T29095C should be C29095T, and T8782C should be C8782T.
Covid-19 is NOT from bats; at least not naturally.
It is claimed that SARS-CoV-2 came from bats, with pangolins acting as an intermediary host. However, this is impossible as the spike proteins of all bat and pangolin coronaviruses have monobasic S1/S2 cleavage sites, whereas, that of SARS-CoV-2 has a multibasic S1/S2 cleavage site. The spike proteins of the human coronaviruses OC43, and HKU1, both have a multibasic cleavage site. This is strong evidence of genetic engineering.
Bat RaTG13 669 GICASYQTQTNS----RSVA 684
SARS-Cov-2 669 GICASYQTQTNSPRRARSVA 688
Above, we compare the S1/S2 cleavage sites of SARS-Cov-2 and the bat virus RaTG13. It has been found that coronavirus spike proteins need to be cleaved/cut in two places in order for them to induce fusion (virus-cell, or cell-cell fusion). These two places are called the S1/S2 and S2' cleavage sites. In the case of SARS-Cov-2 and RaTG13 the S1/S2 cleavage occurs after the bold R and before the S. The bat S1/S2 cleavage motif R is not efficiently cleaved by common enzymes. However, if the arginine amino acid R is replaced by the sequence RRAR then it is well-known, and has been well-known for decades, that the ubiquitously found enzyme, furin, recognizes this sequence/motif and cuts the spike protein very efficiently during the virus' production (in the trans-Golgi-network) by your cells. It has also been known for decades that this greatly increases the pathology of the virus.[22][40][41]
Now SARS-Cov-2 just happens to have the amino-acid sequence PRRA inserted just before the bat S1/S2 cleavage motif R. This just happens to create the S1/S2 multibasic furin cleavage motif RRAR which just happens to dramatically increase the pathology of the bat virus. No similar furin cleavage motif is found in any bat or pangolin virus.
The only other significant difference between the SARS-Cov-2 and RaTG13 spike proteins occurs in the receptor binding domain (RBD). Here the RBD of RaTG13, which does not bind very efficiently to human ACE2-receptors, has been swapped for another RBD, one that just happens to bind extremely well to human ACE2, one we are told is from a pangolin virus. It can be proved that the swapping of the RBDs has been engineered by looking at the ratio of synonymous mutations to non-synonymous mutations within the RBD, and comparing this to the ratio outside the RBD.
The following table compares the S1/S2 and S2' cleavage sites of the SARS-CoV-2 spike protein, with the S1/S2 and S2' cleavage sites of bat viruses spike proteins. It has been copied from [23 Figure 1]. With its furin cleavage motif SARS-CoV-2 is clearly the odd one out.
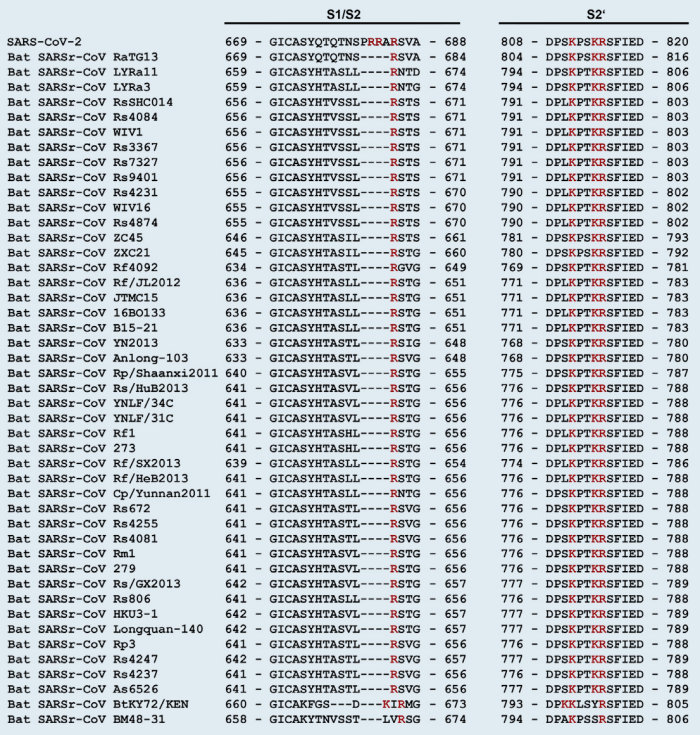
Further evidence that Covid-19 is not natural.
If the amino-acid sequence PRRA was introduced by a recombination event, then the nucleotide sequence coding for it, that is CCT CGG CGG GCA, must have come from some other related virus. However, the segment CGG CGG cannot be found in any coronavirus spike cleavage sequence. In fact, the CGG CGG motif is not found among the spike cleavage sequences of any known virus, strongly indicating that SARS-CoV-2 has been engineered. [42] Two further papers concerning the genetic engineering of SARS-CoV-2 are [49] & [50].
Covid-19 Cover-up in Rome.
The researchers, Bartolini, Bordi, Capobianchi, Carletti, Castilletti, Colavita, Ippolito, Lalle, Nicastri, Rueca, of the National Institute for Infectious Diseases (INMI), in Rome, have put their names to the following three papers/preprints;
[1] Molecular characterization of SARS-CoV-2 from the first case of Covid-19 in Italy.
[2] Virological characterization of the first two Covid-19 patients diagnosed in Italy.
[3] SARS-CoV-2 Phylogenetic Analysis, Lazio Region, Italy, February-March 2020.
Here are some quotes from [1];
"On January 29, 2020, two Chinese spouses (patient 1, female; patient 2, male), coming to Italy as tourists from Hubei province, were hospitalized at the National Institute for Infectious Diseases "L. Spallanzani", Rome, with fever and respiratory symptoms."
This couple were Italy's first Covid-19 cases.
"A virus isolate was obtained from the sputum of patient 1, with cytopathic effects evident 24 h post-inoculation. At the time of writing, virus isolation from the nasopharyngeal swab sample collected from patient 2 was not successful, likely due to the lower viral load, therefore no further analysis was performed on the virus detected in patient 2. Next-generation sequencing was performed on the respiratory samples from patient 1 and on the primary isolate, prior to any further passage, by using the Ion Torrent S5 platform." [1; page 1]
We have the following quotes from [2];
"On January 29th, 2020, two spouses, a 66-year-old woman (Patient 1, Pt1) and a 67-year-old man (Patient 2, Pt2) visiting Rome for vacation, were admitted at INMI, as possible Covid-19 cases. Both patients arrived in Italy on January 23rd from Wuhan, Hubei Province, China, and since January 28th presented relevant respiratory symptoms." [2; page 6]
"First samples collected at diagnosis (nasopharyngeal swabs on both patients and sputum of Pt1) were immediately inoculated into the cell culture for isolation purpose. The follow-up samples (were stored).... Next generation sequencing was performed using Ion Torrent S5 platform as described in [9]." [2; page 5]
Here it is implicit that both the viruses of both Patient 1 and Patient 2 were sequenced. Further, we read;
"The partial Pt2 sequence was very similar to the sequence of Pt1, and consistent with the full genome sequence of the strain isolated by the national reference center (EPI_ISL_412974) from Pt2 nasopharyngeal swab."
This completely contradicts what the INMI authors have previously said, above.
The important point here is that Patient 2's virus was isolated and sequenced. In fact, it was isolated from Patient 2's nasopharyngeal swab. The partial sequences (MT008022 = EPI_ISL_406959; MT008023 = EPI_ISL_406960) of Pt1 and Pt2 are tiny identical 322-base sequences (which were not worth submitting). Their mention is probably just to confuse the meaning of the sentence. In [3] we read;
"We named the sequences INMI3-INMI10 for their detection at National Institute for Infectious Diseases and analyzed them together with the previously published INMI1 and INMI2 [1], along with all the sequences from Italy posted to GISAID database by April 11, 2020." [3; page 1]
INMI1 and INMI2 were the names given to the viruses of Patients 1 and 2. INMI3-INMI10 were the names for the next 8 Covid-19 viruses sequenced by the group. We are told that the INMI group sequenced INMI1 and INMI3 to INMI10 using the Ion Torrent S5 platform. However, instead of sequencing Patient 2's virus, INMI2, themselves, they had it sequenced by the "national reference center," which, of course, may be another reference to themselves, but according to the GISAID database (EPI_ISL_412974) it is the Virology Laboratory of the Army Medical Center. EPI_ISL_412974 is recorded as:
EPI_ISL_412974 2020-01-29 2020-03-01 hCoV-19/Italy/SPL1/2020 Italy/Rome G11083T G26144T
where the format is: id, collection date, submission date, full name, location, and mutations.
So, what was it that caused patient 2's virus, INMI2, to be treated so differently?
One also finds the contradictory statements:
"Full genome sequences of Pt1 were obtained by NGS from both virus isolate and clinical sample (nasopharyngeal swab)." [2; page 7]
"The reads from the two respiratory samples of patient 1 were merged to obtain a better coverage along the virus genome, and in this paper are referred to as data from the clinical sample." [1; page 1]
So what is going on here?
It seems the virus of Patient 1 must have been sufficiently different from that of Patient 2 to call for a cover-up.
After reading these papers it is clear that their purpose was to blame China for the spread of the virus to Italy, even if there was no evidence for this. When the viral sequences of Patients 1 and 2 turned out to be different, this made it unlikely that one of the pair had caught the virus from the other, and actually points to the couple being deliberately infected, which points away from China. So, the differences in the sequences had to be covered up.
Reading further one finds;
"Considering the consensus sequences, two non-synonymous changes with respect to the Wuhan-Hu-1 NCBI Reference Genome were observed in the sequence from the clinical sample from patient 1: G11083T, leading to L3606F change in Orf1a, and G26144T, leading to G251V change in Orf3a. One additional synonymous substitution in Orf1a (A2269T) was detected in the isolate but not in the corresponding clinical sample. All variants were confirmed by Sanger sequencing." [1; page 1]
This is incorrect. This quote implies that the G11083T mutation was found in the isolate sequence of Patient 1. However, the G11083T mutation is nowhere to be found in the isolate sequence.
The isolate sequence from patient 1 is MT066156 = EPI_ISL_410545 = Italy/INMI1-isl/2020.
The clinical sequence from patient 1 is MT077125 = EPI_ISL_410546 = Italy/INMI1-cs/2020.
From the database we have;
EPI_ISL_410545 2020-01-29 2020-02-17 Italy/INMI1-isl/2020 Italy/Rome [isl=isolate]
A2269T G26144T GACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29868-
MT066156 2020-01-30 2020-02-14 ITA/INMI1/2020 Italy
A2269T G26144T GACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29868-
EPI_ISL_410546 2020-01-31 2020-02-17 Italy/INMI1-cs/2020 Italy/Rome [cs=clinical sample]
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAG1- G11083T G26144T TGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29842-
MT077125 2020-01-31 2020-02-17 ITA/INMI1/2020 Italy
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAG1- G11083T G26144T TGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29842-
As you can see, the claimed G11083T mutation is not among the mutations of the isolate. On further investigation it turns out that position 11083 is recorded as indeterminate, i.e., as the letter N. All the other 29,866 bases were successfully determined (as either A,C,G or T). Only this one base, at the important (to this case) position, 11083, was not successfully determined. What are the chances of that? Anyway, this means that certain software will display the (potential) MT066156 mutations as:
MT066156 2020-01-30 2020-02-14 ITA/INMI1/2020 Italy
A2269T G11083N G26144T GACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29868-
where the G11083N is a potential mutation, but, as yet, not determined to be such, and this looks a bit like a G11083T mutation. In any case, a potential mutation seems good enough for these researchers. Of course, in reality, the G11083T mutation was never part of the isolate sequence, and the N had to be added to the isolate sequence to even suggest that it might have been.
In fact, reading between the lines, it seems likely that patient 1's sequence was initially the isolate sequence, and patient 2's was initially the clinical sequence. Something like this:
patient 1's sequence
== the wife's sequence
== isolate sequence/EPI_ISL_410545
== A2269T G26144T GACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA29868-
patient 2's sequence
== the husband's sequence
== EPI_ISL_412974
== G11083T G26144T
== clinical sequence/EPI_ISL_410546 without end mutations
And when it was pointed out that the couple's viral sequences were different, the husband's viral sequence was called a variant of the wife's, and the two were added together to form something new that was also called a clinical sequence, to further confuse the issue. Whether patient 2's sequence was initially the clinical sequence, or not, makes no difference to what has been said above. This is just my guess at what actually happened. In any case, things were done to hide the differences between the couple's viral sequences.
The database shows that Patient 1 and Patient 2 were the only people in all of Italy with viruses belonging to the V clade (i.e., they had at least the mutation G26144T). The nine sequences from Italy in the V clade are clearly just copies of the viral sequences of Patient 1 and Patient 2. All other Italian sequences were clade G (i.e., they had at least the mutation A23403G). This has also been noted here:
"The analysis of sequence data shown in GISAID indicates that the initial introduction of SARS-CoV-2 in Italy through 2 infected tourists in January was effectively contained, and no further circulation of similar clade V strains has been so far detected." [3; page 1]
Note also that the mutation A2269T is only found in these sequences from Rome. It has not been found anywhere else in the world. In particular, it has never been found in China. So when the INMI group claimed that everything was consistent with the couple bringing the virus from China, they were lying.
The Italian sequences plus mutations are listed here. By the way, there is a suspiciously small number of Italian sequences in all the collections.
Train travel tells a story about Covid-19.
It is obvious that the story we are being told about the Covid-19 epidemic is very wrong.
Everyday 59 trains (32 bullet trains and 27 normal trains) leave Wuhan for Beijing.[2]
These trains carry between 600 and 1200 passengers.
Of course, for the period around Chinese New Year, every train would have been packed.
However, let us assume that only 50 passengers on each train from Wuhan end up in Beijing.
So every day 59 * 50 = 2,950 people from Wuhan go to Beijing.
The virus circulated in Wuhan for roughly 40 days before the authorities took action.
Therefore 59 * 50 * 40 = 118,000 people from Wuhan end up in Beijing.
These 118,000 + those arriving by plane + those arriving by automobile, result in 442 confirmed cases of Covid19.
Everyday 155 trains (103 bullet trains and 52 normal trains) leave Wuhan for Changsha (Hunan Province).
Similarly, we have 155 * 50 * 40 = 310,000 people from Wuhan ending up in Changsha.
These 310,000 + those arriving by plane + those arriving by automobile, result in 1,018 confirmed cases of Covid19.
Everyday 127 trains (85 bullet trains and 42 normal trains) leave Wuhan for Zhengzhou (Henan Province).
Similarly, we have 127 * 50 * 40 = 254,000 people from Wuhan ending up in Zhengzhou.
These 254,000 + those arriving by plane + those arriving by automobile, result in 1,273 confirmed cases of Covid19.
And, so on,....
Over the same time period, a few thousand Chinese from Wuhan arrive in Iran.
These are supposedly responsible for 12,729 confirmed cases by March 16.
Over the same time period, a few thousand Chinese from Wuhan arrive in Italy.
These are supposedly responsible for 21,157 confirmed cases by March 16.
Regarding the last two estimates: What I know is that the number of one-way tickets from Wuhan to Paris in the first quarter 2019 was 4,232. Using this 90 day period as a proxy, this is 4,232 * 4/9 = 1,880 passengers for the 40 day period of interest. The "few thousand" passengers from Wuhan to Italy & Iran is a guess based on this. More accurate numbers would be welcome.
Summing up we have:
| More than | 004,000 Chinese from Wuhan visit Hong Kong | resulting in | 00,141 confirmed cases. |
| More than | 082,000 Chinese from Wuhan visit Shanghai | resulting in | 00,353 confirmed cases. |
| More than | 118,000 Chinese from Wuhan visit Beijing | resulting in | 00,442 confirmed cases. |
| More than | 090,000 Chinese from Wuhan visit Chongqing | resulting in | 00,576 confirmed cases. |
| More than | 158,000 Chinese from Wuhan visit Nanjing | resulting in | 00,631 confirmed cases. |
| More than | 310,000 Chinese from Wuhan visit Changsha | resulting in | 01,018 confirmed cases. |
| More than0 | 254,000 Chinese from Wuhan visit Zhengzhou0 | resulting in | 01,273 confirmed cases. |
| More than | 234,000 Chinese from Wuhan visit Guangzhou | resulting in | 01,357 confirmed cases. |
| Less than | 001,000 Chinese from Wuhan visit Iran | resulting in | 12,729 confirmed cases. |
| Less than | 001,000 Chinese from Wuhan visit Italy | resulting in0 | 21,157 confirmed cases. |
The confirmed cases are those reported on March 16, 2020.
How is it visitors from Wuhan to major Chinese cities have an almost zero transmission rate?
Yet we are told that visitors from Wuhan to foreign countries have a very high transmission rate.
How can this be?
Wuhan is in the middle of China.
How could the disease bypass all the major Chinese cities and massively infect huge numbers in Iran and Italy?
Why didn't the disease infect large numbers in the major Chinese cities (before the lockdown)?
Conclusion: The story we are being told about Covid-19 = SARS-CoV-2 is a big lie.
Plane travel tells the same story as Train travel.
We use the one-way bookings from Wuhan to various countries in the first quarter of 2019 to estimate the number of airline passengers.[4] The top ten destinations from Wuhan were:
| Thailand | 74,185 |
| Japan | 29,710 |
| Taiwan | 25,752 |
| Hong Kong 0 | 21,852 |
| Malaysia | 19,105 |
| Korea | 18,623 |
| Australia | 15,020 |
| Cambodia | 13,456 |
| USA | 13,267 |
| Singapore | 12,959 |
Note that neither Iran or Italy are in the top ten destinations.
We are interested in 40 days from the 90 day period, so we multiply by 4/9. We have:
| More than | 32,971 Chinese from Wuhan visit Thailand | resulting in | 0,0827 confirmed cases of Covid19. |
| More than | 13,204 Chinese from Wuhan visit Japan | resulting in | 01,140 confirmed cases of Covid19. |
| More than | 11,445 Chinese from Wuhan visit Taiwan | resulting in | 0,0215 confirmed cases of Covid19. |
| More than | 09,712 Chinese from Wuhan visit Hong Kong 0 | resulting in | 0,0386 confirmed cases of Covid19. |
| More than | 08,491 Chinese from Wuhan visit Malaysia | resulting in | 01,624 confirmed cases of Covid19. |
| More than | 08,277 Chinese from Wuhan visit Korea | resulting in | 09,037 confirmed cases of Covid19. |
| More than | 06,675 Chinese from Wuhan visit Australia | resulting in | 02,044 confirmed cases of Covid19. |
| More than | 05,980 Chinese from Wuhan visit Cambodia | resulting in | 0,0087 confirmed cases of Covid19. |
| More than | 05,896 Chinese from Wuhan visit USA | resulting in0 | 46,450 confirmed cases of Covid19. |
| More than0 | 05,759 Chinese from Wuhan visit Singapore | resulting in | 0,0509 confirmed cases of Covid19. |
The source of this information is the OAG (Air Travel Intelligence) Traffic Analyzer.
Conclusion: The virus was deliberately spread to certain areas. Other areas were forgotten about.
The Covid-19 antibody test.
The easiest to use Covid-19 test looks for antibodies that the body has used to fight the disease. It does not tell how long ago the disease occurred. The assumption is that it MUST BE very recent. But that is only because "everyone" believes that everything started with Wuhan, which it didn't.
Such a test is BioMedomics COVID-19 IgM/IgG Rapid Test: BioMedomics has developed and launched one of the world's first rapid point-of-care lateral flow immunoassays for the diagnosis of coronavirus infection. The test has been used widely by the Chinese CDC (Center for Disease Control and Prevention) to combat infections and is now available globally. This test detects both early marker and late marker, IgM/IgG antibodies in human finger-prick (capillary) or venous whole blood, serum, and plasma samples.... BioMedomics Rapid IgM-IgG Combined Antibody Test for COVID-19 is used to qualitatively detect IgG and IgM antibodies of the novel coronavirus in human serum, plasma or whole blood in vitro. [The U.S. was slow on the uptake, and it was only on March 16 that the FDA recommended this product for healthcare workers at the point-of-care. Previous to this qRT-PCR (quantitative real-time polymerase chain reaction) was used.]
I also came across this pamphlet which states: "Results from the COVID-19 IgG/IgM Rapid Test Cassette (Whole Blood/Serum/Plasma) should not be used as the sole basis to diagnose or exclude SARS-COV-2 infection or to inform infection status."
The pamphlet also states: "The COVID-19 IgG/IgM Rapid Test Cassette.... shows some cross reactivity with samples positive for SARS-CoV antibody and Rheumatoid Factor. It is possible to cross-react with samples positive for MERS-CoV antibody. Positive results may be due to past or present infection with non-SARS-CoV-2 coronavirus strains, such as coronavirus HKU1, NL63, OC43, or 229E."
So, if in the past you have had SARS-CoV-1, MERS-CoV (very unlikely) or one of the common cold corona-viruses HKU1, NL63, OC43, or 229E, (more likely) then you just might test positive for Covid-19.
Corona-viruses have a worldwide distribution, and cause 10-15% of common cold cases. Covid-19 is a corona-virus.
Now, if you have had one of the common cold corona-viruses like HKU1, NL63, OC43, or 229E, and thus test positive for Covid-19, and die of pneumonia, then you will probably be counted as one of those who died of Covid-19, even though you never had the disease.
It is said of the corona-viruses OC43 and 229E: "They are among the viruses that cause the common cold. Both viruses can cause severe lower respiratory tract infections, including pneumonia, in infants, and the elderly," which sounds a lot like the corona-virus Covid-19.
The deliberate spreading of the virus was probably only to China, Iran, and Italy, where a more virulent strain was released. Since the Chinese strain had almost zero transmission rate, it quickly died out. The initial high transmission rate in Wuhan was due to the disease being deliberately spread. Spreading the virus before the Chinese New Year travel extravaganza suggests that the plan was to infect all of China.
The article: "Rates of evolutionary change in viruses: patterns and determinants" states:
For nearly all RNA viruses examined, overall rates of nucleotide substitution fall in the range of 10^-2 to 10^-5 nucleotide substitutions per site, per year (subs/site/year), with most of the viruses exhibiting rates within one order of magnitude of 1 x 10^-3 subs/site/year. For an RNA virus with a genome of 10,000 bases, this is equivalent to the fixation of 10 substitutions per genome, per year.
10^-2 = 0.01, 10^-3 = 0.001, 10^-4 = 0.0001 and 10^-5 = 0.00001.
The nucleotide substitution rate is also known as the evolutionary rate.
Nextstrain.org actually gives you a rate estimate of around 25 substitutions per year. The estimate is hidden under the Clock option. Click the Clock option and it appears on the data graph. 25 substitutions per year seems incredibly wrong when you even have individual genomes with that many substitutions, and that supposedly within a couple of months, not a full year.
25 substitutions per year corresponds to 25/30,000 = 0.0008 = 8 × 10^-4 substitutions per site per year.
Assuming an exponential growth model, this means that after one year, 25 mutations (per genome) will have become fixed in the population. After two years, 625.
The mutation/error rate.
The mutation rate should really be called the error rate as the changes in the RNA sequence are caused by their "duplication" protein not being very accurate. The error/mutation rate is (usually) defined as the average number of errors created in genomes of viral progeny, per base, per replication cycle (mut/nuc/rep). Covid-19 actually has an error checking protein so should have a low error/mutation rate compared to other RNA viruses.
The error/mutation rate can be high but the substitution/evolutionary rate low.
For an error/mutation to count in the evolutionary rate it has to become established in the population which rarely occurs. Typically, a virus with a one nucleotide error/mutation will sit in a sea of billions of viruses without that particular error, and will not become established in the population.
Nextstrain.org
There are a few reasons to worry about nextstrain.org's reporting of the Covid-19 epidemic. The main reason is their giant culls of hundreds of sequences. For example, on April 07, 552 of 3087 genomes just disappeared. Similarly, on April 22, 1902 of 3170 genomes disappeared, while 329 were added. On May 19, 1340 of 5669 genomes disappeared.... So, why were they fudging the data? Were they "cherry picking"?
To try and understand what was happening I listed those sequences kept, and those deleted (in the April 22 cull) in this file. Those kept are in column one, those deleted are in column two. What is notable, is that 93% of the Welsh varieties, 87% of the Scotch varieties, and 68% of the English varieties, just disappeared. What is it that makes the British varieties so unwanted? One thing, is that the British varieties are very different to the standard Wuhan genome (i.e., their Divergence is large). This suggests that the "cherry picking" is done to maintain the fiction that all varieties are from the Wuhan genome.
The Divergence provides a measure of the "closeness" between two genomes (of similar size). It is defined to be the difference in the number of nucleotide mutations between the two. When comparing genomes of different sizes you need to divide the Divergence by the genome length.
Here are a few genomes with large Divergence that appeared on nextstrain, only to soon disappear:
SouthAfrica/RO2606/2020 Divergence=35
Fujian/IM3520004T/2020 Divergence=35
England/20142083004/2020 Divergence=34
Wales/PHWC-24DF0/2020 Divergence=32
England/20144037404/2020 Divergence=30
Columbia/GVI-97181/2020 Divergence=30
DRC/KN-0051/2020 Divergence=30....
There are more here if anyone is interested. All but 5 of these have now been purged. Concerning those 5;
Senegal/640/2020 has had its Divergence changed from 25 to 20 then 19 then 18.
Senegal/328/2020 has had its Divergence changed from 24 to 21 then 18 then 17.
India/nimh-0116/2020 has had its Divergence changed from 22 to 18 then 17.
Luxembourg/LNS0522318/2020 has had its Divergence changed from 21 to 17.
Norway/2093/2020 has had its Divergence changed from 24 to 11.
I would imagine by now that the last five have also disappeared, or been further trimmed.
Concerning nextstrain.org's presentation; I have been asked how a data point can differ from the standard Wuhan genome (that is, have non-zero Divergence), yet report "No nucleotide mutations". The reason is that the missing mutations are reported on the branches leading to the data point in question. If you add up the number of mutations reported on the branches, plus any reported for the data point itself, you get the number of points of difference, that is, the Divergence. I have illustrated this for the data point Senegal/640/2020. You can view the explanatory picture here. An active version can be found here (it seems that the sequence Senegal/640/2020 has disappeared).
{kind=link}
The Senegal/640/2020 sequence is defined by stating its differences to the standard Wuhan genome. The differing nucleotides are [C3037T] [C241T A23403G] [C14408T] [G25563T] [C1059T] [G11083T] [A2825G A2826C T2827A A2831C G29372A C29375G T29377C A29381C G29384T + 2 more]
Their total, that is, the Divergence, is = 1 + 2 + 1 + 1 + 1 + 1 + 11 + 0 = 18.
The term "AA mutations" stands for amino acid mutations. Some nucleotide substitutions do not cause any change in the amino acids produced. Thus, there is no change in the protein sequences, and usually no physical change in the organism. These substitutions are called synonymous.
If you think that the above sequence Wales/PHWC-24DF0/2020 with Divergence of 32 is impressive, here is a Turkish genome with Divergence 60. Comparing it to the standard Wuhan genome, as usual, it is:
C100A T580A G779C T946A T1100G C1101T A1106T A1119C A1134T G1156A G1210A C1225A T1359C G1397A C1420T G1470A C1473T A1475C G2250A C2455T A2475T G2549C T2586A G2591A G2612C G2715T A2932G C3117T G3146C C3787T C4084T C7392T T10532A C11232T G11234A C13476T C13492T C14286T G14310A T14394A C14407A G14430A G14443T T14682G G14710A T14740C C14763A G14773T T14808A C15101A T15119A G15958A C19763A T26396A T26551C C26753T C27103T G28109T T28688C G29742T.
The virus is called hCoV-19/Turkey/6224-Ankara1034/2020 and has GISAID id EPI_ISL_417413.
And here is another monster. This Malaysian virus has Divergence 69.
It is called hCoV-19/Malaysia/186197/2020 and has id MT372483 and GISAID id EPI_ISL_417919.
It is defined by its differences from the standard Wuhan genome, which are:
-320T T2628- G2632T -2633A C2636T A2679C T2737A A10215G A10225C T11104C T12113C A13409G C13424T G13469A T15242A G15416A T15417A T15428A G15429A T15432A G15433A T15437A G15438A C15441A G15449A C15469T G15475A A15478G T15969A -16267G A16270- C16271T T17549C A17553G T17582A G17601A T17618A T17643A C17644A T17647A T17650A T17652A G17667A T17673A C17845T A17871G T17873C C17874T G17887A T22907A A23122T A26945G G26946C C27106T A27107G T27108C -29699G A29700G T29709- T29710A A29812G T29817A A29819T G29825T A29827G -29864AAAAA T29867A G29868A C29870A
Note that this Malaysian branch would have diverged from the Wuhan branch about 1.3 years ago.
There is a very interesting virus from the United States (Wisconsin) that has Divergence 50.
It is called hCoV-19/USA/WI-GMF-00928/2020 and has GISAID id EPI_ISL_426161.
It is defined by its differences from the standard Wuhan genome, which are:
T78G C241T A739G C1059T C2933T C3037T C9866T C12400T C14408T A14747G A20755C A21536T T21537A T21539A A21550T A21551C A23403G C25366T A25367C A25368G C25373G A25377T A25379T G25563T C26447T A26449C G26452T T26453C C26455G C26456T T26457A T26462A C26464G G26466T G26467C G27364T A27365T G27366C C27367G A27369T C27371T A27372C A27373T G27376T A27383T T28238A G28239T T28240C G28242C T28245G
It is mainly of interest because this virus also occurs on nextstrain.org where it has Divergence 12. The mutations presented by nextstrain.org are C241T A739G C1059T C2933T C3037T C9866T C12400T C14408T A14747G A20755C A23403G G25563T which form a subset of the mutations of EPI_ISL_426161 just listed above. They have been marked in red. You can find the nextstrain.org version here.
It would be interesting to hear why the other 38 mutations were thrown away.
There is another very interesting virus from Wuhan China.
It is called hCoV-19/Wuhan/HBCDC-HB-04/2019 and has GISAID id EPI_ISL_412900.
It is defined by its differences from the standard Wuhan genome, which are:
G765C A2215C T4608A T4747C C4763G T6006C T6820C A6837C T6976A T6978A T6980C G6986C T6988A C7006A T9316A G9324A T9325A G9648C G9653C -9825T A9826C T9831C T9861G T21656G A24325G G28597C
It is interesting because it is from Wuhan, because it was acquired on the 30th December 2019, because its Divergence is 26, and because nextstrain.org has not bothered to display it.
So, at the very beginning of the Covid-19 epidemic there was, at Wuhan, a strain of Covid-19 that differed from the standard Wuhan genome by 26 mutations. Now that is super-super-fast evolution.
March 16; Confirmed Cases in the Chinese Provinces.
On March 16, the Johns Hopkins University website
https://www.gisaid.org/epiflu-applications/global-cases-covid-19/
reported the following numbers of cases of Covid-19 in each of the Chinese provinces:
67,794 confirmed cases Hubei China (this province includes Wuhan)
1,357 confirmed cases Guangdong China (this province includes Guangzhou)
1,273 confirmed cases Henan China
1,231 confirmed cases Zhejiang China
1,018 confirmed cases Hunan China
990 confirmed cases Anhui China
935 confirmed cases Jiangxi China
760 confirmed cases Shandong China
631 confirmed cases Jiangsu China
576 confirmed cases Chongqing China
539 confirmed cases Sichuan China
482 confirmed cases Heilongjiang China
442 confirmed cases Beijing China (this province includes Beijing)
353 confirmed cases Shanghai China (this province includes Shanghai)
318 confirmed cases Hebei China
296 confirmed cases Fujian China
252 confirmed cases Guangxi China
245 confirmed cases Shaanxi China
174 confirmed cases Yunnan China
168 confirmed cases Hainan China
146 confirmed cases Guizhou China
141 confirmed cases Hong Kong China
136 confirmed cases Tianjin China
133 confirmed cases Shanxi China
132 confirmed cases Gansu China
125 confirmed cases Liaoning China
93 confirmed cases Jilin China
76 confirmed cases Xinjiang China
75 confirmed cases Inner Mongolia China
75 confirmed cases Ningxia China
18 confirmed cases Qinghai China
10 confirmed cases Macau China
1 confirmed case Tibet China
It has been suggested that the Chinese have lied about the low numbers, but this is obviously false; Why would they give real figures for Wuhan, but false figures for other cities? And, anyway, it would be impossible to keep an on-going epidemic secret, especially in Hong Kong.
The complete Train Schedule for trains from Wuhan.
| High Speed Train Schedule from Wuhan | |||
|---|---|---|---|
| Destination | Trains | Duration | Ticket Fare |
| 1st/2nd Class Seat | |||
| Beijing West | 032 departures from 07:00 to 18:36 | 4h12m - 6h2m | CNY 832.5/520.5 |
| Shanghai | 035 departures from 07:27 to 18:55 | 3h51m - 6h20m | CNY 464.5/289 |
| Hong Kong | 002 departures at 14:20 and 16:00 | 4h38m - 5h2m | CNY 1,082.5/678.5 |
| Xi'an North | 014 departures from 09:17 to 18:05 | 3h55m - 5h20m | CNY 727.5/454.5 |
| Chengdu | 016 departures from 06:25 to 14:06 | 8h00m - 9h57m | CNY 601/375 |
| Chongqing | 038 departures from 06:25 to 16:12 | 5h54m - 7h32m | CNY 419/261.5 |
| Guangzhou | 066 departures from 06:45 to 19:41 | 3h45m - 4h51m | CNY 708.5/443.5 |
| Shenzhen North | 026 departures from 07:25 to 17:49 | 4h17m - 5h38m | CNY 838/538 |
| Futian | 026 departures from 07:25 to 17:49 | 4h17m - 5h38m | CNY 838/538 |
| Hangzhou East | 012 departures from 06:45 to 18:00 | 4h29m - 6h24m | CNY 454/283 |
| Nanjing | 059 departures from 06:45 to 20:04 | 2h30m - 3h56m | CNY 245/153.5 |
| Shenyang | 004 departures from 07:21 to 09:05 | 10h5m - 12h13m | CNY 1,238/767 |
| Zhengzhou | 085 departures from 07:00 to 21:05 | 1h44m - 4h1m | CNY 294.5/184 |
| Harbin West | 001 departure at 07:55 | 12h33m | CNY 1,630.5/1,012.5 |
| Changsha South 0 | 103 departures from 06:45 to 21:19 | 1h18m - 2h2m | CNY 264.5/164.5 |
| Nanning | 007 departures from 07:06 to 14:49 | 6h23m - 7h39m | CNY 753/470.5 |
| Xianning | 058 departures from 06:53 to 21:19 | 0h24m - 1h47m | CNY 62/37 |
| Shiyan | 013 departures from 07:30 to 20:00 0 0 | 1h57m - 2h56m | CNY 225/140 |
| 0 | |||
| Normal Train Timetable from Wuhan | |||
| Destination | Trains | Duration | Ticket Fare |
| Soft/Hard Sleeper | |||
| Beijing | 27 departures from 01:21 to 23:42 | 10h33m - 18h38m | CNY 427.5/279.5 |
| Shanghai | 06 departures from 18:09 to 22:54 | 10h11m - 18h29m | CNY 377.5/247.5 |
| Guangzhou | 34 departures from 00:50 to 23:55 | 10h36m - 18h26m | CNY 443.5/255.5 |
| Xi'an | 11 departures from 00:21 to 23:32 | 10h34m - 15h15m | CNY 357/214 |
| Chengdu | 10 departures from 01:09 to 23:26 | 10h36m - 22h11m | CNY 443.5/272.5 |
| Hangzhou | 08 departures from 12:43 to 22:54 | 8h5m - 15h19m | CNY 346/208 |
| Xiamen | 02 departures at 08:01 and 12:14 | 9h27m - 14h50m | CNY 405.5/277.5 |
| Changsha | 52 departures from 00:19 to 23:55 | 3h13m - 5h17m | CNY 152.5/107.5 |
| Zhengzhou | 42 departures from 00:37 to 23:42 | 4h24m - 9h8m | CNY 242/174 |
| Nanchang | 20 departures from 01:53 to 19:36 | 3h2m - 8h41m | CNY 194.5/142.5 |
| Yichang East | 09 departures from 04:34 to 23:26 | 2h8m - 6h23m | CNY 180.5/134.5 |
| Chongqing North | 07 departures from 04:34 to 22:36 | 7h22m - 18h21m | CNY 346/239 |
| Shenzhen | 07 departures from 05:30 to 21:05 | 12h42m - 18h00m | CNY 467.5/279.5 |
| Tianjin | 08 departures from 01:33 to 22:08 | 11h38m - 20h42m | CNY 443.5/299.5 |
| Guiyang | 05 departures from 00:19 to 23:38 | 15h26m - 17h19m | CNY 456.5/299.5 |
| Shiyan | 09 departures from 00:11 to 23:12 | 4h30m - 7h55m | CNY 204/136 |
| Kunming | 03 departures from 00:19 to 23:38 | 22h53m - 28h43m | CNY 630/397 |
| Harbin West | 03 departures from 07:33 to 18:04 | 23h16m - 37h44m 0 | CNY 749.5/491.5 |
The Last Update of the Schedule was on Jan 10, 2020.[3]
Interesting videos.
Here is an interesting video from two Californian doctors who recommend that the state's lockdown be lifted immediately. The main speaker mixes up his statistics somewhat, and is inconsistent in his application of them. Also, he appears to be recommending exposure to dangerous pathogens as a way to built up one's immune system, however, he is actually recommending exposure to less virulent strains (which he assumes exist) to do this. Well worth watching (even if they have been heavily pushed by the false opposition (which usually pushes somewhat suspect material)).
https://www.youtube.com/watch?v=36AyI7mwZdk (censored)
https://www.youtube.com/watch?v=xfLVxx_lBLU (censored)
https://www.youtube.com/watch?v=UaTYYk3HxOc (censored)
https://www.youtube.com/watch?v=vJprwe_rWeM (censored) and here 73M.
I recommend the following video by Dr Yeadon (former Pfizer Chief Science Officer). You can watch it here or download it here (640x360 70M).
At one point Dr Yeadon states that the epidemic is over. Now, if the epidemic was a naturally occurring event, he would be correct. However, who knows what evil those spreading the virus may yet have in store for the world.
Here is a video concerning a father and son who both had major reactions to the covid vaccine. It is claimed that both had (the disease) Covid-19 immediately before taking the vaccine. Both developed multiple blood clots. The son, mainly in his brain, and the father, mainly in his lungs. The father permanently lost the use of a quarter of his left lung. The son may eventually recover. More on this below.
Rand Paul has had enough of lockdowns, etc. Rand Paul chooses freedom. See this historic video 13M.
In this video Erin Marie Olszewski is interviewed regarding her time at Elmhurst Hospital in New York. From a review of her book Undercover Epicenter Nurse we have, "Worse, people who had tested negative multiple times for Covid-19 were being labeled as Covid-confirmed and put on Covid-only floors (where they caught the disease). Put on ventilators and drugged up with sedatives, these patients quickly deteriorated; even though they did not have coronavirus when they checked in." Once on ventilators, they invariably died. In the video she calls it murder. You can download the video from here 640x360 96M. The interview was on May 12, 2020.
Erin records a conversation between nurses, and doctors, where the nurses refuse to kill a patient by letting him die when he could have been saved. Apparently, the doctors had been instructed to treat patients as DNR (do not resuscitate) even though they were not DNR. The nurses refused to go along with this. You may ask why the media has refused to investigate her claims. You may ask why the media has refused to even report them.
"Do not attempt resuscitation" orders were also applied to care home patients throughout England, even though neither the patient, nor their family, had signed a "do not resuscitate" document. Doctors who killed patients this way say they were just following orders, and the pressure of the pandemic is used as an excuse for the policy. You may ask why the media has told you nothing about this.
In this video Erin Olszewski is interviewed by thehighwire.com 640x360 38M.
An unexplained correlation between vaccinations and Covid-19 deaths.
This short video graphically shows the correlation. If anyone knows why the vaccinations appear to cause Covid-19 deaths, let me know. I can guess, but I don't know.
References. Part 1.
[1] The Washington Post has said: "All domestic and international flights out of Wuhan were canceled since the city was placed under lockdown last week to prevent the spread of the virus. But the city government says 5 million people left the city before the quarantine came into effect, and some apparently want to return to their homes." and "The mayor of Wuhan said last weekend that 5 million people had left the city before a lockdown was imposed."
[2] https://www.travelchinaguide.com/china-trains/beijing-wuhan.htm "There are over 55 pairs of trains running between Beijing West Railway Station and Wuhan/Hankou Railway Station. Among them, over 30 pairs are bullet trains with the journey time of 4.5-6 hours and another 25 or so pairs are normal ones which take 10-18 hours for the whole way."
[3] https://www.travelchinaguide.com/china-trains/wuhan-schedule.htm
[4] https://www.oag.com/blog/2019-ncov-tracking-down-the-bug
[5] 601 RNA sequences from Iceland have been submitted to GISAID, but nextstrain.org shows only 21 (they used to show more). I looked at 560 of these Icelandic sequences and found 231 to be distinct.
[6] This article states; "Several studies already report that roughly 50% of the infections are asymptomatic (i.e, no symptoms at all), which makes the early detection and isolation of the infected problematic."
[7] This article states; "Notably, the 2019-nCoV strains were less genetically similar to SARS-CoV (about 79%) and MERS-CoV (about 50%)."
[8] The rate is from this article. Note that the rate is not for the whole SARS virus but for just over two thirds of it (the ORF 1ab segment which is about 20,000 bases long). I have since came across this article which gives estimates from 0.80 to 2.38 × 10^-3. The upper estimate here translates to about 70 new varieties fixed in a year, rather than 12. Anyway, 12 or 70, makes little difference to the thrust of the argument above. Nextstrain.org actually gives you a rate estimate of about 25 substitutions per year, but never explains exactly what this means. For more on viral evolution click here.
Note: RNA is like DNA. It is the type of genetic code used by the virus.
Part 2. The Covid-19 Vaccines.
Pfizer-BioNTech and Moderna's Covid-19 mRNA Vaccines.
The general idea of these mRNA vaccines is to have your own cells produce the Covid spike protein. From the site of production (the endoplasmic reticulum) the protein is transported to, and anchored in the cell wall (plasma membrane). There the protein is exposed to the surrounding fluid where it can be investigated by your immune system. Once your immune system recognizes the danger, your body will attempt to block, or kill, everything that expresses the spike protein. In particular, since the Covid virus (SARS-CoV-2) expresses the spike protein, your immune system will attempt to block, or kill it, whenever it finds it.
Obviously, there is much missing from this statement.
What triggers the immune system?
On the viral surface three spike proteins join together to form a single structure called a spike. There are about 26 of these spikes scattered over the surface of the virus (where they are anchored in the viral membrane). Now the vaccine leads to such spikes on the surface of your cells (where they are anchored in the plasma membrane). If these spikes just lay about on the cell surface, then why would this trigger your immune system? If they do nothing, how will your immune system recognize them as dangerous?
Generally, cells have to die, and unexpectedly, before the their debris is sifted by the immune system to find what killed them. If your body finds something unusual among the debris it invites further investigation by specialist cells that determine whether this unusual thing is to be flagged as dangerous, and whether, in the future, anything expressing this unusual thing is to be killed on sight. Thus, to trigger the immune system, the spikes on the cell surface would have to kill a few of your cells, and a few spikes, or bits of them, would have to be found among the debris at the crime scene, so that "what did it" can be established.
Now these vaccine spikes are created alone. They have no help from the sibling proteins produced when the Covid virus infects a cell. So can these spikes, completely separated from the rest of the virus, trigger your immune system? Can they kill anything? We read;
Severe cases of Covid-19 are associated with extensive lung damage and the presence of infected multinucleated syncytial pneumocytes. The viral and cellular mechanisms regulating the formation of these syncytia are not well understood. Here, we show that SARS-CoV-2-infected cells express the Spike protein (S) at their surface and fuse with ACE2-positive neighboring cells. Expression of the Spike protein without any other viral proteins triggers syncytia formation. [1]
Like other fusion proteins that are active pH independently, S protein mediates not only fusion between the viral and the cellular membranes during particle entry but also fusion of infected cells with uninfected cells. This process is mediated by newly synthesized S protein accumulating at the cell surface. The resulting syncytia are giant cells containing at least three, often many more nuclei. Cell-cell fusion is used by viruses such as human immunodeficiency virus (HIV), measles virus (MV), or herpes virus to spread in a particle-independent way. The resulting syncytia are documented as pathological consequence detectable in various tissues such as the lung (measles virus), skin (herpes virus), or lymphoid tissues (HIV). In the brain, cell-to-cell transmission via hyperfusogenic F proteins (the measles spike protein) constitutes a hallmark of MV-caused encephalitis as a fatal consequence of acute MV infections manifesting years later (from one day to 15 years). [2]
Note that the spike protein is often simply called the S protein. Syncytia are large multi-nucleate cells formed from the fusion of many cells. That the spike proteins of many coronaviruses, by themselves, can induce syncytia, has been known for decades. We have this from a 2003 review of the molecular biology of coronaviruses:
During infection by some corona-viruses a fraction of the S protein that has not been assembled into virions ultimately reaches the plasma membrane. At the cell surface S protein can cause the fusion of an infected cell with adjacent, uninfected cells, leading to the formation of large, multinucleate syncytia. This enables the spread of infection independent of the action of extracellular virus, thereby providing some measure of escape from immune surveillance. Expression of S without any other viral proteins triggers syncytia formation. [3]
Wild-type Covid spikes have the ability to reach out, grab a neighboring ACE2-positive cell, and cause both cells to merge. This can happen multiple times so that one ends up with large non-functional cells (syncytia) which die. If this happens, then all the individual cells are seen to die unexpectedly, and spikes, and bits of spike, end up among the debris. This shows that the expression of wild-type Covid spikes, by themselves, should trigger an immune response. That is, if a vaccine induces wild-type Covid spikes on your cell's surfaces, then this will trigger an immune response.
Even though the syncytia formed here are non-functional, there are fully-functional syncytia, e.g., in muscles, and the syncytium layer surrounding the placenta.
In vaccine development, the antigen you are trying to invoke an immune response to, often does not have the innate killing power of the spike protein. For example, you might try to invoke an immune response to a small piece of the spike protein known as the Receptor Binding Domain (RBD), which by itself cannot kill cells. In this case you need to add another chemical, called an adjuvant, to kill a few cells, and frame the antigen (the RBD) for the crime. If successful the immune system is triggered, and the antigen targeted for destruction.
To see that the expression of the complete spike protein on the surface of the cell is the "Holy Grail" of Covid-19 vaccine design, we have this:
Vaccine development against the SARS-CoV-2 virus focuses on the principal target of the neutralizing immune response, the spike (S) glycoprotein. Adenovirus-vectored vaccines offer an effective platform for the delivery of viral antigen, but it is important for the generation of neutralizing antibodies that they produce appropriately processed and assembled viral antigen that mimics that observed on the SARS-CoV-2 virus. Here, we describe the structure, conformation and glycosylation of the S protein derived from the adenovirus-vectored ChAdOx1 nCoV-19/AZD1222 vaccine. We demonstrate native-like post-translational processing and assembly, and reveal the expression of S proteins on the surface of cells adopting the trimeric pre-fusion conformation. The data presented here confirms the use of ChAdOx1 adenovirus vectors as a leading platform technology for SARS-CoV-2 vaccines. [4]
It is alarming that the aim of mRNA vaccines is to have the spike protein expressed on the surface of your cells, as this is appears quite dangerous.
How dangerous is the spike protein?
It has been realized that the coronavirus S protein is a type I viral fusion protein with functional similarities to the fusion proteins of phylogenetically distant RNA viruses such as influenza virus, HIV, and Ebola virus. [3]
That the covid virus spike proteins are related to those of the influenza, HIV, Ebola, and SARS-CoV viruses, definitely suggests that they could be quite dangerous. We have:
The data reveal a strong membrane fusion activity of S protein and demonstrate syncytia formation even at undetectable levels of the S protein. [2]
The wild-type Covid spike protein is so strongly fusogenic, that it is capable of producing syncytia even when present in undetectable quantities. These syncytia are strongly associated with severe cases of Covid-19.
Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced Covid-19 pathology. [5]
In summary, Covid-19 is a unique interstitial pneumonia with extensive lung thrombosis, long-term persistence of viral replication in pneumocytes and endothelial cells, along with the presence of infected cellular syncytia in the lung. We propose that several of the Covid-19 disease features are due to the persistence of virus-infected cells in the lungs of the infected individuals for the duration of the disease. [5]
This pronounced ability to fuse cells is typical of coronaviruses in general. A particularly virulent coronavirus, the JHM strain of mouse hepatitis virus, is associated with large syncytia in the brains of mice.
The S protein of the highly virulent coronavirus, mouse hepatitis virus (MHV) strain 4 (JHM), has been shown to exist in a particularly metastable configuration. This results in a hair-trigger spike so highly fusogenic that it can mediate fusion between infected cells and (neighboring) cells lacking receptors, thereby leading to more extensive neuropathogenesis than occurs with other MHV strains. [3]
Natural questions are: Can the Covid virus infect human brains? And, if so, can the spike protein alone do similar damage? The answer to the first question is yes.
Together, these results are the first to show the direct impact that the SARS-CoV-2 spike protein could have on brain endothelial cells; thereby offering a plausible explanation for the neurological consequences seen in Covid-19 patients. [6]
Is the spike protein always dangerous?
By itself, the Covid spike protein should be less dangerous than the virus. For many the Covid virus causes little harm, but for others it is fatal. So for many, the Covid spike protein, by itself, will cause little harm, but for others it may prove fatal. So why not cripple it in some way, while retaining its original conformation? This appears to be what Pfizer-BioNTech and Moderna have done, although they do not claim this.
The Pfizer-BioNTech and Moderna mRNA vaccines produce a spike protein (denoted S-2P) with two amino acid changes from the wild-type. The two changes, K986P and V987P, are to stabilize the pre-fusion form of the protein. This stabilization is necessary to make sure that antibodies are made against the pre-fusion, and not the post-fusion form. A 1967 trial of an RSV (Respiratory Syncytial Virus) vaccine was a disaster as the vaccine induced antibodies that bound to the post-fusion form of the RSV-spike protein, but did not prevent infection, which lead to inflammation, clogged airways, and more severe disease than with no vaccine at all. [7][8]
The idea of stabilizing the pre-fusion form comes from previous work on HIV, RSV, SARS and MERS. The equivalent changes, V1060P and L1061P, that stabilize the pre-fusion form of the MERS spike protein are claimed to significantly impair cell fusion. So, it is probable that the changes, K986P and V987P, do likewise for the Covid spike protein.
The phrase "stabilizing the pre-fusion form" implies that the pre-fusion form can still change to the post-fusion form, that is, that the Covid spike-2P protein can still cause cell fusion, but probably not to the same extent. One guesses that syncytia are still formed, but they are fewer and smaller. Still, it seems that enough cells die to trigger an immune response without an adjuvant.
We have this regarding the MERS S-2P protein:
The introduction of two consecutive proline substitutions at the beginning of the central helix (our 2P design) presents a general approach to produce soluble prefusion coronavirus S ectodomains and overcomes the first hurdle in subunit vaccine development. Due to restricted backbone torsion angles, proline substitution can disfavor the refolding of the linker between the central helix and HR1, which for class I fusion proteins is a key step in the transition to the postfusion conformation. The rigidity of the helix-loop-helix afforded by the prolines impairs or (perhaps) abolishes the membrane fusion activity of the S protein, as evidenced in Fig. 2A. [9]
Actually, membrane fusion is not abolished by the 2P changes, but by this change, that was hiding in the small print:
The S1/S2 furin-recognition site 748-RSVR-751 was mutated to ASVG to produce a single-chain S0 protein. [9]
This mutates the MERS-spike multibasic cleavage site (furin-recognition site). Since it appears that the mutation of the multibasic cleavage site, and the 2P changes, make the vaccine much less dangerous, it is a worry that many vaccines do not include these changes. We have;
The ChAdOx-based vaccine candidate developed by AstraZeneca, as well as the CanSino- and Gamaleya-vectored candidates, use a wild-type version of the spike protein. The same is of course true for the inactivated vaccines produced by Sinovac and Sinopharm. Moderna's and Pfizer's mRNA vaccines are based on a spike construct that includes the PP mutations but features a wild-type cleavage site. [21]
Why is Covid-19 more dangerous to some than others?
The present studies suggest that coinfection of SARS-CoV with some other non- or low-pathogenic respiratory agents, such as Chlamydia, mycoplasma, or bacteria, results in severe lung disease, which is attributed to the proteases produced by the infection with those non-SARS-CoV agents, as has been shown by the enhancement of respiratory diseases caused by influenza virus coinfected with non-pathogenic bacteria. Studies are in progress to see whether coinfection exacerbates pneumonia in mice infected with SARS-CoV. [10]
The above statement concerns SARS-CoV-1. This suggests it may be true for Covid-19. So, if you are unlucky enough to have one of these respiratory infections, when struck by Covid-19, you might die. This would be because these other infections induce the production of certain surface proteases which prime the Covid spike protein, and enable it to form syncytia, that is fuse cells, that is, turn some part of your lungs to mush. This would also probably be true for vaccine spikes.
So, is this also true for covid-19? Little research seems to have been done. We do have:
As with some other RNA viruses, co-infection or activation of latent bacterial infections along with pre-existing health conditions in Covid-19 disease may be important in determining a fatal disease course.... Although preliminary, Mycoplasma pneumoniae has been identified in Covid-19 disease, and the severity of some signs and symptoms in progressive Covid-19 patients could be due, in part, to Mycoplasma or other bacterial infections. [13]
Altogether, our analysis described the distribution of SARS-CoV-2-infected cells in patient's BAL and revealed the presence of a viral co-infection by the human metapneumovirus that dampens the immune activation of the monocyte compartment in the infected patient. Further large-scale analyses of mild versus severe patients need to be conducted to better understand if the co-infection is correlated or even causative in SARS-CoV-2 pathology. [14]
As noted above: Studies are in progress to see whether coinfection exacerbates pneumonia in mice infected with SARS-CoV. [10]
I have found one of these studies. In it the authors state that co-infection of mice with SARS-CoV, and Pasteurella pneumotropica (a low-pathogenic bacterium) induces severe pneumonia resembling that found in human SARS cases (> 35% mortality rate). Mice infected with either pathogen alone, do not develop severe disease. [16]
These studies were initiated due to the previous discovery that elastase, like TMPRSS2, enables SARS-CoV to enter directly from the cell surface, and that this direct entry likely leads to serious disease.[17][10] Since a Pasteurella pneumotropica infection is known to elicit elastase production in the lungs, it was predicted that co-infection with SARS-CoV would lead to severe pneumonia, which it did. This is important, as neutrophils, a type of immune cell, secrete elastase into the lungs during infection. This suggests that coinfection of SARS-CoV with any respiratory agent that causes inflammation is likely to lead to more serious disease.
So, what about Covid-19? Astonishingly, the research into Covid-19, and elastase, seems not to have been done. We do have:
Neutrophil elastase was an independent predictor of Covid-19 lung damage. [18]
This says that the presence of elastase is highly correlated with severe lung damage. Given this, how is it possible that necessary studies have not been done? The same article states;
Many arguments suggest that neutrophils could play a prominent role in Covid-19. However, the role of key components of neutrophil innate immunity in severe forms of Covid-19 has (received) insufficient attention. [18]
And, what about vaccine spikes? The research has not been done. All one can say is, if you have any condition that leads to inflammation of your lungs, you should probably not receive the vaccine, as it will likely lead to greater cell death, due to increased formation of syncytia in your lungs.
Here is a video concerning a father and son who both had major reactions to the covid vaccine. It is claimed that both had (the disease) Covid-19 immediately before taking the vaccine. Both developed multiple blood clots. The son, mainly in his brain, and the father, mainly in his lungs. The father permanently lost the use of a quarter of his left lung. The son may eventually recover. The video can be found here or here.
Leaving aside the inexplicable lack of research, one wonders about the cytokine storm that is often blamed for the observed lung damage. Does severe lung damage cause a heightened immune response that results in a cytokine storm, or does a faulty immune response cause a cytokine storm that results in severe lung damage? Since it is obvious that severe lung damage will cause a cytokine storm, what is the evidence, that the observed cytokine storm actually causes the lung damage.
In order to proceed we need to develop a little background knowledge.
How does the Covid-19 virus get into your cells?
The Covid-19 virus can enter a cell in two different ways, by membrane fusion and endocytosis. The following diagrams illustrate this.
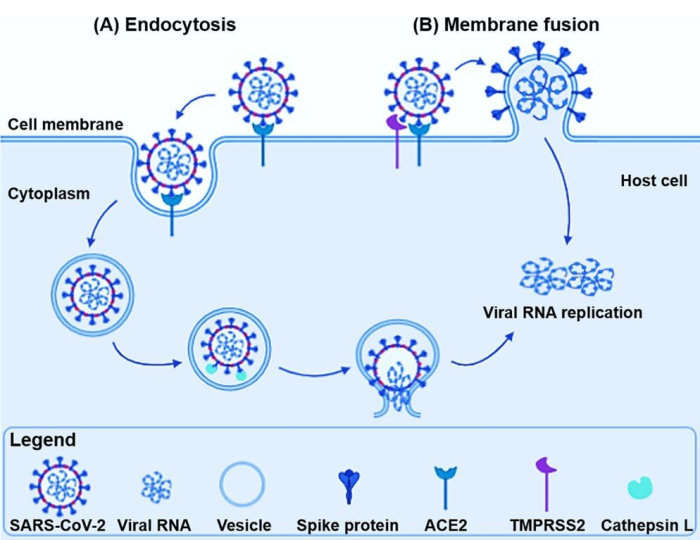
In membrane fusion the Covid virus spike protein attaches to an ACE2 receptor of the target cell, then, if present on the target cell, a TMPRSS2 receptor cleaves the S2 fusion sub-protein. This primes the spike protein for fusion. On fusion, the viral membrane and cell wall merge, which dumps the viral RNA into the cytoplasm. In the absence of TMPRSS2 receptors, the virus travels by the endocytosis pathway. That is, once the virus has attached to an ACE2 receptor, it is wrapped in a membrane and bought inside the cell. The membrane plus engulfed material is called an endosome. Inside the endosome are various proteins that cutup other proteins. One of these, cathepsin L, primes the spike protein for fusion. On fusion, the viral and endosome membranes merge. Again, this results in the viral RNA being dumped into the cytoplasm.
It has been established that the Covid virus mainly uses membrane fusion to enter cells. As you can see from the diagram, membrane fusion leaves copies of the spike protein on the cell surface, although this fact has been obscured by having the spikes of the rightmost viral particle, drawn differently. Here is another diagram sketching Covid-19 virus replication:
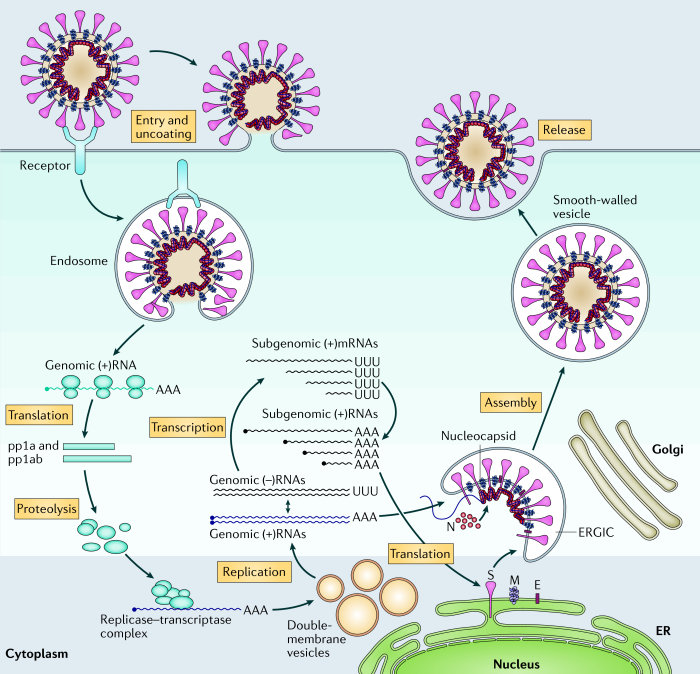
Again, the membrane fusion pathway has been obscured.
Why do the vaccines work at all?
I've come across a problem that I have not been able to resolve. Basically, the problem is;
It is believed that a Covid infection begins in the epithelial cells of the respiratory tract. Nasotracheal epithelial cells, and lung alveolar epithelial cells are known to carry both ACE2 and TMPRSS2 receptors. ACE2 has been found to colocalize with TMPRSS2 in alveolar epithelial type II cells, so that after binding, the virus can immediately break into the cell. So, we expect to see rapid expansion of infected sites due to cell-cell and virus-cell fusion.
We also know that the antibodies, etc, created by vaccination, are in the blood and interstitial fluid, which is separated from the action. So, how do the antibodies, etc, stop the infection? It seems that by the time they find out about the infection it will be too late.
So, why would the covid vaccines work, at all?

In the diagram the gap between the alveolus and capillary (the interstitium) is wider than it should be.
It is believed that a measles infection begins in the epithelial cells of the respiratory tract, and can infect alveolar epithelial cells resulting in pneumonia, as can Covid-19. We also know that there is an effective measles vaccine that produces antibodies, etc, in the blood and interstitial fluid. This is separated from the scene of the action (within the alveoli) by the epithelial basement membranes, yet this vaccine works. So, how does it work, and what might this tell us about Covid-19?
The paper "Cell-to-Cell Contact and Nectin-4 Govern Spread of Measles Virus from Primary Human Myeloid Cells to Primary Human Airway Epithelial Cells," may provide some answers. It says:
For many years, measles virus was thought to enter through the apical surface (the surface in contact with the air) of airway epithelial cells, a misconception based on studies performed with polarized immortalized cell lines. Using well-differentiated primary cultures of airway epithelial cells from human donors, we demonstrated that measles virus has a clear preference for basolateral (the surface anchored to the body) entry. [15]
So, the measles virus may not infect the alveoli from the air, after all. We also have:
Measles is a highly contagious, acute viral illness. Immune cells within the airways are likely first targets of infection, and these cells traffic measles virus to lymph nodes for amplification and subsequent systemic dissemination. Infected immune cells are thought to return the virus to the airways. [15]
So, the measles virus first infects cells in the airways, including various immune cells. These infected immune cells spread the virus throughout the body. In particular, they infect the alveolar epithelial cells of the lungs via their basolateral surfaces. So, strange as it may seem, the infection spreads to the alveolar epithelial cells from within the body, not from the air, as one has been told. This would certainly explain why the measles vaccine works.
Perhaps something like this happens with Covid-19. There is a lot of speculation here. The first thing that would need to be checked, is; Can the Covid virus infect immune cells? The answer to this appears to be yes. I have found a paper that states;
SARS-CoV-2 exhibits a polarity of infection in airway epithelium only from the apical membrane; [19]
I am suspicious of this (and other similar reports). Why would the apical membrane (the surface of the cell exposed to the air) have ACE2 receptors? Why would the apical membrane have any receptors at all? What use would they be? And, even if there were a few, wouldn't the surfactant layer prevent them from working? If the world made any sense, then the receptors would be in the basolateral membrane where they would be used to communicate with cells in the rest of the body. The only point of having receptors in the apical membrane would be to allow airborne viruses to break into your body.
Remember, a similar result was claimed for the measles virus.
These results also support the conclusion that measles virus entry occurs selectively at the apical plasma membrane. [20]
However, this has been shown to be false, as mentioned above.
Do mRNA vaccine nanoparticles get into your brain?
So where exactly do the mRNA vaccine nanoparticles go after they have been injected into your arm?
The 2017 paper Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses, [30] tells you where the nanoparticles end up when administered to mice. It tells you the concentration of nanoparticles in the various tissues, which indicates how much spike protein will be produced, which indicates how much damage the spike protein will do to these tissues. The numbers below are from Table 1 of the paper. They represent the maximum concentration of nanoparticles in the various tissues (ng/ml) after having received the jab.
| Bone Marrow | 3.35 |
|---|---|
| Brain | 0.43 |
| Cecum | 0.89 |
| Colon | 1.11 |
| Close Lymph Nodes | 2120.00 |
| Distant Lymph Nodes | 177.00 |
| Heart | 0.80 |
| Ileum | 3.54 |
| Jejunum | 0.33 |
| Kidney | 1.31 |
| Liver | 47.20 |
| Lung | 1.82 |
| Muscle (Injection Site) | 5680.00 |
| Ovaries | ???? |
| Plasma | 5.47 |
| Rectum | 1.03 |
| Spleen | 86.90 |
| Stomach | 0.63 |
| Testes | 2.37 |
The study uses Moderna's lipid nanoparticles.
The fact that the payload of these nanoparticles is RNA from influenza, and not covid-19, will make little difference to the results, as (by design) the mRNA is buried in the nanoparticle, below a lipid layer. The mRNA only becomes exposed after a nanoparticle merges with one of your cells, and dumps its mRNA into that cell. Note that the lymph nodes close to the injection site receive an extremely high dose of nanoparticles. This means that the generated spike protein will do significant damage to them. What are the long term effects of this damage?
This study investigated both male, and female mice, but only reported on the male mice. This lends credence to the rumors that a dangerously large quantity of the nanoparticles ends up in the ovaries. One leaked report, supposedly from Pfizer, puts the concentration of nanoparticles in the ovaries at about half of that found in the spleen, which is a lot. This has caused much speculation. Is the vaccine designed to cause female sterility? However, such "leaked reports" are often "released" specifically to mislead people. But, who knows? At this point nothing would surprise me.
Vesicular stomatitis virus vaccines.
The vesicular stomatitis virus (VSV) mainly infects animals, but can produce a flu-like illness in humans. VSV has become a favorite for vaccine development as its genome is relatively simple, well understood, and easy to manipulate. Vaccines based on VSV are being developed for influenza, HIV, SARS, MERS, Ebola, Marburg, and Lassa fever. The Ebola vaccine, Ervebo, has been trialled and approved for humans. [32-39]
An important feature of the virus is that the native spike protein, called G, can be easily replaced by foreign spike proteins. Now, if you wished to develop a vaccine against SARS-CoV-2 you would replace the RNA-code for G by the RNA-code for the SARS-CoV-2 spike protein (in the vesicular stomatitis virus genome). Then, after a certain amount of routine tweaking you would bring the lifeless RNA to life. The virus so formed will have a vesicular stomatitis virus body (called a backbone or shell) with SARS-CoV-2 spikes protruding from it. This chimeric virus will be new to nature, and if well-designed will be able to reproduce itself (replication-competent). Such viruses can also be engineered to undergo only one round of infection (replication-deficient).
As the only SARS-CoV-2 part of this new virus is the spike protein, it is likely safer than the SARS-CoV-2 virus itself. If this new virus proves to be relatively safe, then it can be administered as a vaccine. In its action this virus is much like the vaccines of Pfizer and Moderna, the main difference being that the delivery vehicle of one is a nanoparticle, and of the other is a hybrid virus. Both methods deliver the genetic code into the cytoplasm where the cellular machinery will crank out SARS-CoV-2 spike protein. Both allow the SARS-CoV-2 spike protein to leak to the cell surface where it invokes an immune reaction. Apparently, this leakage is due to a sub-optimal endoplasmic reticulum retrieval signal (-KxHxx-COOH) in the spike protein's cytoplasmic tail.[27] The hybrid virus will be much more selective in the type of cells it infects as infection is controlled by the SARS-CoV-2 spike protein. Thus it will mainly infect cells expressing ACE2, whereas, nanoparticles will infect any cells they come across.
Of course, this vaccine production strategy has already been carried out for SARS-CoV-2, and a number of vaccine candidates have been engineered. In 2020 Merck began clinical trials of two VSV-type vaccines targeting SARS-CoV-2, but abandoned them in January 2021.
As the hybrid virus VSV-SARS-CoV-2 resulting from swapping-in the wild-type SARS-CoV-2 spike protein was found to replicate poorly, and was pathogenic (significant syncytia formation) the virus had to be serially passaged (cultured in cells for a number of generations) to look for mutations that replicated better, and were less dangerous.
In [31] the researchers passaged the VSV-SARS-CoV-2 virus until two mutations of the spike protein became dominant. One, R685G, was a mutation of the multibasic S1/S2 cleavage site which left the site non-functional. As is always the case, the loss of function of the multibasic S1/S2 cleavage site leads to a virus that out-competes, and in short time dominates, its parental variety. (Incidentally, this sort of thing is evidence that most of these multibasic cleavage sites have been genetically engineered.) The other was a mutation introducing a stop codon at position 1250, leading to the truncation of 24 amino acids at the cytoplasmic tail. This truncation removes most, if not all, of the trafficking signals for the spike protein. Thus more spike protein ends up on the plasma membrane where it is incorporated into new virus particles (VSV-like particles are assembled at the plasma membrane) and if the multibasic S1/S2 cleavage site is still functional, this will also significantly increase syncytia formation. New virus with the R685G mutation, and 21-amino-acid deletion was chosen as their vaccine candidate.
Spike swapping in VSV has long been used to study viral spike proteins. It has become routine and in the paper "A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells," [23] they produce six spike swapped viruses all of which could have become vaccine candidates.
In this paper the researchers create four mutant SARS-CoV-2 spike proteins. They are;
1) SARS-2-S(RaTG) which has the SARS-CoV-2 S1/S2 cleavage site replaced by the bat RaTG13 S1/S2 cleavage site;
2) SARS-2-S(SARS) which has the SARS-CoV-2 S1/S2 cleavage site replaced by the SARS-CoV S1/S2 cleavage site;
3) SARS-2-S(delta) has all the basic amino acids in the S1/S2 cleavage site removed or replaced; and
4) SARS-2-S(opt) which has extra basic amino acids (arginine and lysine) to make the cleavage site super-basic.
They also create two mutant SARS-CoV spike proteins. They are;
1) SARS-S(RaTG) which has the SARS-CoV S1/S2 cleavage site replaced by the bat RaTG13 S1/S2 cleavage site; and
2) SARS-S(SARS-2) which has the SARS-CoV S1/S2 cleavage site replaced by the SARS-CoV-2 S1/S2 cleavage site.
They then incorporate each of these foreign spike proteins into a VSV backbone, creating six different vaccine candidates/viruses. They also introduce the three wild-type spike proteins of SARS-CoV, SARS-CoV-2, and MERS, into a VSV backbone, creating three more vaccine candidates/viruses. Finally they simply produce spikeless VSV particles (i.e., the spike protein G is deleted with no replacement). They do this to investigate the pathology of the nine different spike proteins once expressed in your cells. The spikeless VSV particles (empty vector) serve as a control. Having created these hybrid viruses they then infect cells with them and record the results.
The researchers chose Vero E6 cells (ATCC Cat# CRL-1586) a cell line from monkeys that expresses ACE2 and is commonly used to model human cells. Generally, ACE2 expression in human cells is low.[28][29] They prepared;
1) Vero E6 cells without any added protease/enzymes,
2) Vero E6 cells with extra-cellular trypsin, and
3) Vero E6 cells expressing TMPRSS2.
Each of the nine VSV hybrids, as well as the spikeless particles, were used to infect each of the three cell cultures. This gave thirty cultures to report on. The results are shown as a matrix of small photos (shown below) of the cultures 48 hours after infection. These photos show the results of having the various spike proteins expressed on the surface of the Vero E6 cells. They indicate the level of damage done by the various spike proteins to the ACE2 expressing cells in your body.
For example, the photo in the (1,5) position shows the damage done to Vero E6 cells by being vaccinated with the VSV-SARS-CoV-2 hybrid. It indicates the damage done to your ACE2 expressing cells by having your cells express wild-type SARS-CoV-2 spike protein. It also indicates the damage done by vaccination with any vaccine that uses wild-type SARS-CoV-2 spike protein to invoke an immune reaction. This would include the current covid vaccines as they express wild-type, or almost wild-type, spike protein. As syncytia formation is evident, there will be some tissue damage, but you should recover. The level of adverse side-effects will be high.
The photo in the (3,5) position indicates the damage done to cells expressing both ACE2, and TMPRSS2 by wild-type SARS-CoV-2 spike protein. ACE2 and TMPRSS2 are co-expressed in a small percentage of cells in your lungs, nose, cornea, esophagus, ileum, colon, gallbladder and bile duct. Here, syncytia formation is widespread. This is indicative of significant tissue damage in your lungs, etc. Fortunately, the percentage of nanoparticles reaching your lungs is low, however, it is much higher for your lymph nodes, spleen and liver. [30 Table 1]
The photo in the (3,9) position indicates the damage done by the MERS spike protein to your cells expressing TMPRSS2. Note that MERS uses DPP4, and not ACE2, for viral entry. This illustrates how dangerous MERS is. It can turn your lungs to mush.
A very dangerous Covid-19 virus variant.
The SARS-2-S(opt) hybrid virus looks amazingly dangerous. The relevant photos are (1,1) (1,2) and (1,3). Here, syncytia formation is extreme. Here the cleavage motif at the S1/S2 cleavage site has been destabilized by adding extra arginine (R) and lysine (K) units. In other coronaviruses this causes the spike proteins to be so unstable that they are able to fuse neighboring cells without the need of any receptor. In case you are wondering, this is an example of "gain of function" research.
Fortunately, such heavily multibasic motifs can never establish themselves naturally. Such motifs cause the spike protein to be quite unstable, and thus very dangerous. However, being so unstable the spike fires prematurely, and also participates in cell-cell fusion, rather than being incorporated into new virus. This means that viruses with such motifs replicate poorly, and are no match for the parental-type without such motifs. Viruses having such spikes are seen to be strongly selected against. Thus whenever a mutation occurs that leads to a heavily multibasic cleavage site you have only one non-competitive virus in a sea of viruses that out compete it. Thus it, and its meager offspring, die out by natural selection. This is illustrated by the experiments carried out by the same researchers where they found a nine-fold reduction in the ability of the SARS-2-S(opt) virus to infect TMPRSS2- Vero cells and a seven-fold reduction in its ability to infect human TMPRSS2+ Calu-3 cells. See [23 Figure 2E]. Many other papers have noted similar reductions in fitness for viruses with heavily multibasic cleavage motifs. In fact, this is even true for SARS-CoV-2 with its sub-optimal furin multibasic cleavage site. See, for example, [31][25].
How does the Covid-19 virus reproduce?
How is the vaccine mRNA delivered into your cells?
Are lipid nanoparticles safe?
How do your cells produce the spike protein?
How does the spike protein reach the cell surface?
The first dose primes your body to kill. The second dose labels cells to be killed. Is this good?
What is antibody-dependent enhancement?
Conclusions.
Expression of the spike protein (without any other viral proteins) triggers syncytia formation.
All the currently available Covid vaccines rely on expression of the spike protein to induce an immune response.
Therefore all the currently available Covid vaccines are potentially dangerous.
I would be very wary of all Covid vaccines, especially those where the spike protein retains the multibasic cleavage site, and does not have the 2P changes. All vaccines that use the wild-type spike protein should look at incorporating the 2P changes (K986P and V987P). Removal, or mutation, of the multibasic S1/S2 cleavage site is a must. [21][25] We have:
mutation of the multibasic site completely prevents syncytia formation. [22]
Moreover, deletion of the multibasic motif resulted in a spike protein that was no longer able to induce syncytium formation even in the presence of trypsin or TMPRSS2. [23]
Novavax has such a vaccine candidate. [24]
At this point I would decline the vaccine. I would take a "wait and see" approach. Experimental evidence suggests the vaccines will kill a large number (billions) of your cells. Clinical evidence shows that most will recover from this without much difficulty. However, we also know that the number of adverse events is much, much greater than for the flu-vaccines. Since Covid is not that dangerous to most people, those not at risk should not be vaccinated until a truly safe vaccine is developed.
A number of medical experts say that the current crop of covid vaccines should not be on the market, at all.
A review article on ivermectin concludes that: Moderate-certainty evidence finds that large reductions in Covid-19 deaths are possible using ivermectin. [43] Dr Pierre Kory's testimony before the U.S. Senate, makes a powerful statement as to the effectiveness of ivermectin in treating Covid-19. You can find a few testimonials (from 2020) as to the potency of ivermectin in treating severe covid-19, here. Those that blocked the use of ivermectin and hydroxychloroquine should be shot. The latest ivermectin trial shows a 90% reduction in mortality if used as a prophylactic.
Both hydroxychloroquine and ivermectin have been conclusively shown to be of great benefit to covid-19 patients. A meta-study of 302 hydroxychloroquine trials can be found here. A meta-study of 71 ivermectin trials can be found here.
One of the benefits of "waiting and seeing", rather than being guinea-pigs in a massive experiment run by evil people, is that you get to find out that the vaccine does not work all that well. It also causes many severe adverse events, and you really should not be vaccinated until a truly safe vaccine is developed. That it isn't working well is shown by the talk of "breakthrough cases". For example;
The Massachusetts Department of Public Health tracked a cumulative 9,969 confirmed Covid-19 infections among those fully vaccinated in Massachusetts to date and 106 of those have died. From here.
(By the end of July, 2021) at least 125,000 fully vaccinated Americans have tested positive for Covid and 1,400 of those have died, according to data collected by NBC News. The data is from 38 states; 12 states provided no, or incomplete information, and were not included in the survey.
Of course, the liars spin the vaccines as a great success, despite the breakthrough case deaths.
Given what is now known, no one should be given this vaccine:
1) It doesn't work as claimed.
2) The breakthrough cases are probably ADE (antibody dependent enhancement) showing itself. This means that the vaccine actually increases the risk of catching Covid-19, and can increase the severity of the disease when you encounter the disease in the future.
3) With effective cures available, the disease is simply not dangerous enough to warrant the risk that comes with the vaccine. The disease has been made to look very dangerous by having it deliberately spread in nursing/care homes, and hospitals, where having Covid-19 was truly dangerous.
If you are still contemplating the vaccine, watch this video first. In it an eminently qualified doctor warns of the very real dangers of the covid vaccines. Here is another interview with the same doctor. And another (about half way through).
On October 2, 2021, Dr Peter McCullough spoke at the 78th Annual Meeting of the "Association of American Physicians and Surgeons". Here is a (must watch) video where he shows that the covid-19 vaccines have been proven unsafe, and should have been withdrawn back in February 2021, when only 27 million Americans had been vaccinated. 98M. You can watch a larger version of the video here.
Recently, Dr Peter McCullough appeared on an episode of The Joe Rogan Experience and discussed a range of topics concerning the Covid-19 pandemic. You can download the video from here. 259 M.
This video, from May 2021, is one of Peter McCullough's best interviews. 133M. Here is part one of an August 2021 interview titled "This Interview Could Save Your Life." 64M. Here is part two. 116M.
Dr McCullough certainly gets around. In this video he discuses Covid-19 with Alex Jones. 83M. And here is his March 10, 2021, testimony before the Texas-Senate-HHS-Committee. 25M.
Robert F Kennedy's book The Real Anthony Fauci; Bill Gates Big Pharma and the Global war on Democracy and Public Health. The first couple of hundred pages summarize (one part of) the Covid fraud, and Fauci's leading role in it.
Scott Atlas served as a member of the White House Coronavirus Task Force and Special Advisor to President Trump. In his book he relates an insider's view of events at the White House.
This is just my two pennies worth. Make of it what you will.
References. Part 2.
[1] Syncytia formation by SARS-CoV-2-infected cells.
[2] Quantitative assays reveal cell fusion at minimal levels of SARS-CoV-2 spike protein and fusion from without.
[3] The molecular biology of Coronaviruses.
[4] Native-like SARS-CoV-2 spike glycoprotein expressed by ChAdOx1 nCoV-19/AZD1222 vaccine.
[5] Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced Covid-19 pathology.
[6] The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood–brain barrier.
[7] A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism.
[8] Brief History and Characterization of Enhanced Respiratory Syncytial Virus Disease.
[9] Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen.
[10] Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection.
[11] Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses.
[12] Neuroinvasion of SARS-CoV-2 in human and mouse brain.
[13] COVID-19 Coronavirus: Is Infection along with Mycoplasma or Other Bacteria Linked to Progression to a Lethal Outcome?
[14] Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients.
[15] Cell-to-Cell Contact and Nectin-4 Govern Spread of Measles Virus from Primary Human Myeloid Cells to Primary Human Airway Epithelial Cells.
[16] Co-infection of respiratory bacterium with severe acute respiratory syndrome coronavirus induces an exacerbated pneumonia in mice.
[17] Entry from the Cell Surface of Severe Acute Respiratory Syndrome Coronavirus with Cleaved S Protein as Revealed by Pseudotype Virus Bearing Cleaved S Protein.
[18] Elastase and exacerbation of neutrophil innate immunity are involved in multi-visceral manifestations of Covid-19.
[19] Long-Term Modeling of SARS-CoV-2 Infection of In Vitro Cultured Polarized Human Airway Epithelium.
[20] Entry and Release of Measles Virus Are Polarized in Epithelial Cells.
[21] Introduction of Two Prolines and Removal of the Polybasic Cleavage Site Lead to Higher Efficacy of a Recombinant Spike-Based SARS-CoV-2 Vaccine in the Mouse Model
[22] Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion.
[23] A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells.
[24] Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate.
[25] Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis.
[27] Sequences in the cytoplasmic tail of SARS-CoV-2 spike facilitate syncytia formation.
[28] SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes.
[29] The scRNA-seq expression profiling of the receptor ACE2 and the cellular protease TMPRSS2 reveals human organs susceptible to COVID-19 infection.
[30] Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses.
[31] A single dose of recombinant VSV-ΔG-spike vaccine provides protection against SARS-CoV-2 challenge.
[32] Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses.
[33] A system for functional analysis of Ebola virus glycoprotein.
[34] Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses.
[35] Recombinant vesicular stomatitis viruses from DNA.
[36] Vaccination with Recombinant RNA Replicon Particles Protects Chickens from H5N1 Highly Pathogenic Avian Influenza Virus.
[37] Development of an HIV vaccine using a vesicular stomatitis virus vector expressing designer HIV-1 envelope glycoproteins to enhance humoral responses.
[38] A recombinant VSV-vectored MERS-CoV vaccine induces neutralizing antibody and T cell responses in rhesus monkeys after single dose immunization.
[39] Vesicular Stomatitis Virus-Vectored Multi-Antigen Tuberculosis Vaccine Limits Bacterial Proliferation in Mice following a Single Intranasal Dose.
[40] Major Concerns on the Identification of Bat Coronavirus Strain RaTG13 and Quality of Related Nature Paper.
[41] The Abnormal Nature of the Fecal Swab Sample used for NGS Analysis of RaTG13 Genome Sequence Imposes a Question on the Correctness of the RaTG13 Sequence
[42] SARS-CoV-2 and the secret of the furin site.
[43] Ivermectin for Prevention and Treatment of COVID-19 Infection: A Systematic Review, Meta-analysis, and Trial Sequential Analysis to Inform Clinical Guidelines.
[44] The potential for antibody-dependent enhancement of SARS-CoV-2 infection: Translational implications for vaccine development.
[45] Anti-Severe Acute Respiratory Syndrome Coronavirus Spike Antibodies Trigger Infection of Human Immune Cells via a pH- and Cysteine Protease-Independent FcγR Pathway.
[46] At the Intersection Between SARS-CoV-2, Macrophages and the Adaptive Immune Response: A Key Role for Antibody-Dependent Pathogenesis But Not Enhancement of Infection in COVID-19.
[47] Strictly regular use of ivermectin as prophylaxis for COVID-19 leads to a 90% reduction in COVID-19 mortality rate,...
[48] Why are we vaccinating children against COVID-19?
[49] Should we discount the laboratory origin of COVID-19?
[50] The genetic structure of SARS-CoV-2 does not rule out a laboratory origin.
[51] Chloroquine is a potent inhibitor of SARS coronavirus infection and spread.
[52] In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine.
[53] Hydroxychloroquine: From Malaria to Autoimmunity.
[54] Evaluation of immunomodulators, interferons and known in vitro SARS-CoV inhibitors for inhibition of SARS-CoV replication in BALB/c mice.
[55] Hydroxychloroquine and Chloroquine Retinopathy. D.J. Browning.
[56] Structural basis of receptor recognition by SARS-CoV-2.
[57] Effect of High vs Low Doses of Chloroquine Diphosphate as Adjunctive Therapy for Patients Hospitalized With SARS-CoV-2 Infection. A Randomized Clinical Trial.
[58] Pre-trial questioning. One question being; Why were research subjects exposed to a lethal dose?
Suggestions for improvement can be made here.
Vaccine Adverse Effects.
The U.S. Government collects reports of adverse health events that follow the administration of a vaccine. The database of reported adverse events is called the VAERS (Vaccine Adverse Event Reporting System) database. The raw data can be downloaded from two sites, both of which present the data in ways designed to restrict your access to meaningful information. You can download the data from:
https://vaers.hhs.gov/data/datasets.html
Here the data has been split into three .csv files; 2021VAERSDATA.csv 2021VAERSSYMPTOMS.csv and 2021VAERSVAX.csv in a way that makes the data essentially useless, unless repackaged. This is deliberate.
The separate files make it very difficult to associate particular symptoms with particular vaccines. There are also problems where one file contradicts another, but this is not obvious as the data has been split. An example is; person 1033762 has "Death" listed among symptoms in 2021VAERSSYMPTOMS.csv but she is not listed as having died in 2021VAERSDATA.csv.
You can also download the data from here as a 767MB Microsoft Access database file. Offering a .mdb file restricts access to those having the necessary Microsoft software. The size of the file is also meant to put you off.
I have repackaged the three .csv files as a single text file. You can download it from here (data till 16 Dec. 2022; WinZip opens .gz files). I have only repackaged the data I thought important. I have not included the number of days from the vaccination date to the onset date, which can be calculated from the other data.
If you wish to browse the database go to https://medalerts.org/vaersdb/
You can also try https://wonder.cdc.gov/vaers.html
If you just wish a general summary of the data follow this link. N.B. This includes many foreign cases.
The following statistics report adverse reactions that have occurred within the United States, and have been recorded in the VAERS database. The under-reporting factor is unknown, but is likely to be high.
As of 16 Dec. 2022 there have been 15,615 deaths in the U.S. associated with the three vaccines given to Americans. We have:
| U.S. DEATHS ASSOCIATED WITH COVID-19 VACCINES: | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Date | 09-04 | 07-05 | 04-06 | 02-07 | 30-07 | 27-08 | 24-09 | 22-10 | 19-11 | 17-12 | 14-01 | 11-02 | 11-03 | 08-04 | 06-05 | 03-06 | 01-07 | 29-07 | 26-08 | 23-09 | 21-10 | 18-11 | 16-12 |
| Pfizer | 1128 | 1673 | 1942 | 2238 | 2480 | 2766 | 3253 | 3671 | 4123 | 4357 | 4620 | 4938 | 5326 | 5679 | 5901 | 6057 | 6204 | 6348 | 6496 | 6652 | 6767 | 6930 | 7069 |
| Moderna | 1246 | 1921 | 2216 | 2484 | 2675 | 2865 | 3169 | 3451 | 3767 | 3951 | 4186 | 4506 | 4823 | 5166 | 5366 | 5498 | 5636 | 5801 | 5947 | 6130 | 6249 | 6491 | 6640 |
| Janssen | 72 | 294 | 393 | 486 | 542 | 621 | 746 | 887 | 1029 | 1097 | 1276 | 1367 | 1475 | 1535 | 1563 | 1600 | 1629 | 1659 | 1680 | 1730 | 1762 | 1803 | 1845 |
| Total | 2462 | 3908 | 4574 | 5232 | 5724 | 6280 | 7199 | 8044 | 8957 | 9443 | 10121 | 10853 | 11668 | 12429 | 12879 | 13205 | 13522 | 13862 | 14179 | 14569 | 14837 | 15283 | 15615 |
| MYOCARDITIS: | |||||||||||||||||||||||
| Date | 09-04 | 07-05 | 04-06 | 02-07 | 30-07 | 27-08 | 24-09 | 22-10 | 19-11 | 17-12 | 14-01 | 11-02 | 11-03 | 08-04 | 06-05 | 03-06 | 01-07 | 29-07 | 26-08 | 23-09 | 21-10 | 18-11 | 16-12 |
| Pfizer | 23 | 62 | 371 | 737 | 880 | 969 | 1139 | 1260 | 1339 | 1427 | 1533 | 1643 | 1696 | 1729 | 1754 | 1772 | 1788 | 1804 | 1819 | 1836 | 1848 | 1866 | 1875 |
| Moderna | 32 | 54 | 212 | 353 | 421 | 458 | 534 | 613 | 674 | 719 | 789 | 859 | 876 | 893 | 908 | 915 | 924 | 931 | 935 | 942 | 963 | 973 | 984 |
| Janssen | 0 | 4 | 13 | 22 | 29 | 34 | 41 | 52 | 62 | 66 | 78 | 83 | 87 | 90 | 91 | 92 | 93 | 94 | 97 | 98 | 100 | 102 | 104 |
| PERICARDITIS: | |||||||||||||||||||||||
| Date | 09-04 | 07-05 | 04-06 | 02-07 | 30-07 | 27-08 | 24-09 | 22-10 | 19-11 | 17-12 | 14-01 | 11-02 | 11-03 | 08-04 | 06-05 | 03-06 | 01-07 | 29-07 | 26-08 | 23-09 | 21-10 | 18-11 | 16-12 |
| Pfizer | 38 | 69 | 200 | 420 | 503 | 559 | 664 | 766 | 856 | 910 | 973 | 1024 | 1053 | 1078 | 1099 | 1119 | 1122 | 1131 | 1147 | 1166 | 1181 | 1192 | 1198 |
| Moderna | 30 | 57 | 150 | 260 | 311 | 352 | 404 | 458 | 515 | 559 | 601 | 633 | 660 | 680 | 699 | 705 | 710 | 716 | 722 | 728 | 735 | 739 | 747 |
| Janssen | 0 | 13 | 22 | 34 | 49 | 57 | 67 | 78 | 88 | 90 | 96 | 100 | 103 | 107 | 109 | 111 | 111 | 111 | 113 | 115 | 115 | 116 | 119 |
| PULMONARY EMBOLISM: | |||||||||||||||||||||||
| Date | 09-04 | 07-05 | 04-06 | 02-07 | 30-07 | 27-08 | 24-09 | 22-10 | 19-11 | 17-12 | 14-01 | 11-02 | 11-03 | 08-04 | 06-05 | 03-06 | 01-07 | 29-07 | 26-08 | 23-09 | 21-10 | 18-11 | 16-12 |
| Pfizer | 91 | 310 | 645 | 752 | 808 | 899 | 993 | 1080 | 1186 | 1260 | 1340 | 1417 | 1456 | 1493 | 1519 | 1541 | 1557 | 1574 | 1591 | 1601 | 1610 | 1626 | 1636 |
| Moderna | 107 | 341 | 663 | 786 | 859 | 917 | 986 | 1048 | 1104 | 1162 | 1227 | 1298 | 1339 | 1360 | 1376 | 1394 | 1400 | 1414 | 1427 | 1450 | 1462 | 1471 | 1478 |
| Janssen | 11 | 246 | 339 | 384 | 408 | 436 | 469 | 506 | 546 | 559 | 577 | 596 | 617 | 620 | 625 | 634 | 636 | 637 | 641 | 645 | 646 | 647 | 652 |
| THROMBOCYTOPENIA: | |||||||||||||||||||||||
| Date | 09-04 | 07-05 | 04-06 | 02-07 | 30-07 | 27-08 | 24-09 | 22-10 | 19-11 | 17-12 | 14-01 | 11-02 | 11-03 | 08-04 | 06-05 | 03-06 | 01-07 | 29-07 | 26-08 | 23-09 | 21-10 | 18-11 | 16-12 |
| Pfizer | 66 | 128 | 247 | 311 | 355 | 373 | 397 | 417 | 458 | 484 | 513 | 553 | 579 | 591 | 599 | 611 | 618 | 629 | 633 | 647 | 655 | 664 | 673 |
| Moderna | 65 | 120 | 218 | 269 | 284 | 299 | 320 | 334 | 357 | 367 | 385 | 404 | 418 | 430 | 438 | 448 | 453 | 460 | 466 | 471 | 478 | 482 | 484 |
| Janssen | 9 | 92 | 122 | 137 | 147 | 152 | 164 | 184 | 197 | 205 | 214 | 219 | 232 | 235 | 235 | 237 | 239 | 240 | 243 | 245 | 247 | 249 | 250 |
The above are monthly (28 day months) starting 9 April 2021. Weekly results can be found here. I was hoping that someone would use the weekly data to determine the extent of the throttling that has occurred.
The under-reporting factor has not been accurately determined. It has been estimated to be as low as 5 and as high as 40. This means that the numbers presented above need to be multiplied by this factor. Consequently, the numbers presented above are a huge under-estimate of the true number of adverse events.
As their Nov 26, 2021 "update", the evil VAERS people simply provided the Nov 19 data again. By "accident", of course.
The VAERS people delay all reports by one to two weeks (but some are delayed by months) so they can weed out those adverse events they feel are not directly related to the vaccines. Yet we have report 1180840 where the person involved died due to a traffic accident. How did this pass VAERS censorship? Clearly, the VAERS people have chosen to let the report stand. They may have even submitted the report themselves. They have done this for propaganda purposes. It has been done to stress that not all adverse events are actually caused by the vaccines, implying that you should heavily discount the numbers. However, they have already removed reports they feel are not directly related to the vaccines, so the numbers have already been heavily discounted. They do this to deceive you. That they delete hundreds of reports is clear from the numerous gaps in the VAERS ID. Actually, a lot of the gaps are due to foreign reports.
Other popular topics are:
Proof that Adolf Hitler was a double agent.
A few books on Religion and Mythology (Free Downloads).
A large collection of photos of Jewish political leaders wearing skullcaps, e.g., Nelson Mandela, Tony Blair, Gordon Brown, etc.
There are no ancient Jewish cities in Israel, but there are lots of ancient Greek cities.
WOW; Presidents Reagan, Clinton, Bush, Obama, Trump, and Biden are Jews.
Is it too late to save Florida from Drowning? The Conspiracy to hide Global Warming.
The free Operating System Linux running Microsoft Operating Systems on Virtual Computers (a DIY guide).
Proof that the name ADAM is of Greek origin.
You can leave a note at the forum
http://preearth.net/imagemagick/viewtopic.php?f=3&t=5
No need to join. Just leave a message.